
Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review


1
State Key Laboratory of Shale Oil and Gas Enrichment Mechanism and Effective Development, Beijing 100101, China
2
Institute of Unconventional Oil & Gas, Northeast Petroleum University, Daqing 163318, China
3
Department of Applied Chemistry, Federal University Dutsin-Ma, Dutsin-Ma P.M.B. 5001, Nigeria
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(1), 34; https://doi.org/10.3390/molecules27010034
Submission received: 27 November 2021
/
Revised: 15 December 2021
/
Accepted: 17 December 2021
/
Published: 22 December 2021
(This article belongs to the Special Issue Applications of Stable Isotope Analysis)
Abstract
:Over time, stable isotopes have proven to be a useful tool in petroleum geochemistry. However, there is currently insufficient literature on stable isotope geochemistry of the organic elements within shales and crude oils in many petroleum systems around the world. As a result, this paper critically reviews the early and recent trends in stable isotope geochemistry of organic elements in shales and crude oils. The bulk and compound-specific stable isotopes of H, C, and S, as well as their uses as source facies, depositional environments, thermal maturity, geological age, and oil–oil and oil–source rock correlation studies, are all taken into account. The applications of the stable isotopes of H and C in gas exploration are also discussed. Then, the experimental and instrumental approaches to the stable isotopes of H, C, and S, are discussed.
1. Introduction
The analysis of stable isotopes of various elements present in the organic matter (OM) enables us to understand diverse geochemical processes that take place during geological periods [1]. Stable isotopes have been used in gas and petroleum exploration for correlation [1,2,3,4,5,6,7,8], maturation [9,10,11,12], and OM evolution studies [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19], as well as assessing the depositional environment of the source rocks [13,14,15,16,17,18,19]. Because OM is made up mostly of carbon (C) and hydrogen (H), with other heteroatoms, i.e., nitrogen (N), oxygen (O), and sulfur (S), knowing the stable isotopes of these elements is crucial for petroleum exploration [1]. However, the analysis of isotopic compositions of oxygen and nitrogen has been rarely employed in petroleum exploration, mainly due to analytical challenges in quantitatively converting organic oxygen and nitrogen to CO2 and N2 [1].
To understand geochemical and geological processes, stable isotope geochemistry uses the ratios of isotopes between distinct phases or chemical species in a compound [20]. Isotopes of an element are atoms with the same number of protons but a different number of neutrons, resulting in a different atomic mass [20]. In contrast to unstable or radioactive isotopes, stable isotopes have nuclei that do not decay over time. Thus, except for physical, chemical, and biological processes that lead to their fractionation, stable isotope abundances in geologic materials remain constant across time [20]. The ratio of two stable isotopes of the same element can be used to calculate the relative amounts of light and heavy isotopes in a phase or chemical species. The degree to which the light or heavy isotope is preferentially integrated into a substance during a phase transition or chemical reaction can be determined by comparing isotope ratios of materials [1,20]. Although, temperature, reaction kinetics, and mass can all influence the degree of this preferential incorporation, knowing such ratios can provide us with insight into reaction mechanisms, formation temperatures, and other information regarding the evolving earth [1,20,21].
C and H stable isotopes, since they are the most abundant elements, are important for determining the processes that sedimentary organic matter has undergone. Carbon and hydrogen, for example, both have two stable isotopes: 12C and 13C and 1H and 2D. 12C makes up to 98.899 wt.% of the whole carbon pool, while 13C makes up only 1.111 wt.% [22]. The complete hydrogen pool is made up of 99.985 wt% of 1H and 0.0105 wt% of 2D [22]. Silverman et al. [2], conducted the first in-depth isotopic investigation of petroleum. The initial basic understanding of petroleum isotope geochemistry was developed by [3,4,5,23,24], in the United States, [25,26] in East Germany, [27] in Italy, [28] in West Germany, and [6,29,30], in the Soviet Union. In addition, in the 1960s, the Galimov group at Gubkin’s Institute conducted theoretical and practical research of isotope fractionation in hydrocarbon systems, as well as oil and gas studies in the Volga–Ural, Sakhalin, and Kaspian regions, which are some of the most pioneering studies in this field reported in the literature [31,32,33,34,35,36,37,38,39,40].
Sulfur is a common element in nature that plays a key role in a variety of natural processes, and understanding sulfur isotopic composition sheds light on a wide range of geological events throughout history [41,42,43]. Sulfate is an important link between the carbon, sulfur, and iron cycles, particularly in sedimentary basins, through geochemical processes such as bacterial sulfate reduction (BSR), iron sulfide production, and organic matter sulfation [7,8,44,45]. Bacterial sulfate reduction (BSR) is an anaerobic process that occurs in anoxic conditions with abundant organic matter as a reductant and results in a depletion of 34S in sulfide products (such as pyrite) compared to coeval carbonate-associated sulfate if no obvious alteration occurs in the burial process [46,47,48].
Even though there are many papers on the stable isotope geochemistry of C, H, and S and their applications to hydrocarbon explorations, the literature is still insufficient when considering the importance of C, H, and S isotopes in oil and gas explorations. As a result, the early and modern uses of the stable isotope geochemistry of organic elements (C, H, S) within shales and crude oils are further explored in this review paper.
2. Standards and Notation
The stable isotope delta (δ) notation, which is expressed by the following equation, is the measured values relative to an internationally accepted standard.
δ(‰) = [(Rsample − Rstandard)/Rstandard] × 1000
R depicts the isotope abundance ratio, such as 13C/12C, 18O/16O, 34S/32S, 15N/14N, or D/H (2H/1H). For example, the δ value for carbon is a convenient way to characterize tiny differences in the relative abundance of 13C in biological matter. In comparison to the standard, a negative value indicates that the sample is depleted in the heavy isotope, whereas the positive value indicates that the sample is enriched in the heavy isotope. To express relative isotope composition, the terms “light” and “heavy” are replaced for carbon, and instead “13C-depleted” and “13C-enriched” are used, respectively. The isotopic ratios of stable isotopes and their corresponding reference standard are shown in Table 1 [21].
3. Stable Isotope Fractionation
Isotope fractionation is a physical phenomenon that occurs when mass differences between isotopes produce variations in relative abundance [49]. Isotopic fractionation occurs in nature as a result of binding strength during chemical, biological, and physical processes [20]. Isotopes that are “heavy” make stronger bonds than isotopes that are “light”. As a result of isotopic fractionation associated with these processes, the isotopic composition of crude oils and sediments, as well as their constituents, can be affected by isotopic source signatures and processes such as maturation and biodegradation [50,51,52]. Kinetics and equilibrium are the two fractionation processes that cause these isotopic shifts. While both processes may be active, the kinetic isotope impact is the most relevant in most hydrocarbon exploration investigations, such as in natural gas exploration studies [52,53,54].
Kinetic Isotope Effect (KIE)
Different reaction rates for heavy vs. light isotopes would cause KIE related to degradation and maturation processes (e.g., 13C vs. 12C). The presence of a “heavy” species (e.g., 13C) in the degradation-targeted bond raises the bond cleavage activation energy and, as a result, slows the rate of degradation. The leftover substrate becomes increasingly enriched in “heavier” isotope species as degradation progresses (i.e., 13C/12C increases). Isotope effects associated with degradation are significant for the atoms present when the chemical bond is broken (primary isotope effects), whereas secondary isotope effects are moderate for the remaining atoms. Considering hydrogen, only secondary isotope effects are significant at the normal precision of CSIA (compound-specific isotopic analysis). When a biodegradation process exclusively targets a single atomic position (or positions) (e.g., oxidation of a methyl group in toluene), the overall expression of isotope fractionation, as assessed by CSIA, is “diluted”. The intrinsic role of an isotope that affects the atoms directly involved in the reaction is unaffected by molecule size and can be determined using CSIA data [20,21,37,38,55]. The initial rate-limiting phase of the reaction cycle is reflected by isotope effects on biodegradation. In other circumstances, the delayed step may not be a bond cleavage but rather a non-isotope fractionating process, such as the development of the substrate–enzyme complex. There will be no isotope fractionation if the latter is entirely rate limiting. The rule of thumb is that biodegradation isotope effects are restricted to tiny compounds that can easily pass through degraders’ cell membranes. This would include chlorinated ethenes and ethanes, methyl tertiary butyl ether (MTBE), and mono-aromatic compounds that have been extensively researched and where isotope fractionation has been seen [55,56]. Fractionation has been seen in the lighter hydrocarbons in the C5 to C10 range in exploration studies [57]. Semi-volatile environmental pollutants including long-chain n-alkanes, multi-ring polycyclic aromatic hydrocarbons (PAHs), and polychlorinated biphenyls (PCBs) often do not produce measurable isotopic fractionation during biodegradation. It is also worth noting that in larger compounds with more than 10–12 carbon atoms, the isotopic alterations that occur to the one carbon atom that is being attacked are diluted by the presence of the other carbon atoms that are not impacted. As a result, overall changes in the isotopic makeup of these bigger molecules are usually undetectable when isotope values are measured [21]. Isotope fractionation can also be caused by physical processes such as phase transitions, sorption, and diffusion [58,59,60]. Different kinetic energies of gaseous phase molecules result from the mass difference between isotope species, resulting in differential rates of vapor migration and different bond energies of light isotope- vs. heavy isotope-substituted molecules, affecting phase partitioning equilibria and evaporation–condensation [20,21]
4. Experimental and Instrumental Methods in Stable Isotopes (C, H, and S)
The analytical methods applied in the analyses of stable carbon, hydrogen, and sulfur isotopes are reported below.
4.1. Stable Isotopic Analyses
4.1.1. Fractionation of Sediment Extracts and Crude Oils
Column chromatography with silica gel–alumina as the stationary phase is generally used to fractionate extracts and crude oils. A typical glass column is 50 cm long and has an internal diameter of 0.5 cm. The column is rinsed twice, with DCM first and then light petroleum spirit (petroleum ether). After that, the column is filled with n-hexane and cotton wool is used as a resting pad for the stationary phase, silica gel (SiO2). After that, the stationary phase (SiO2) is added. Two (2 g) of Alumina (Al2O3) is used to stabilize the surface. A total of 70 mL of n-hexane, 70 mL of dichloromethane–n-hexane (2:1, v/v), and 70 mL of dichloromethane–methanol (1:1, v/v) are used to elute the saturated, aromatic hydrocarbon, and polar fractions, respectively. Each fraction is recovered by carefully evaporating solvents on a rotary evaporator, followed by the removal of the leftover solvent under a nitrogen gas stream (modified from [61]). The recovered saturated hydrocarbon, aromatic hydrocarbon, and polar fractions can now be analyzed using elemental analysis–isotope ratio mass spectrometry (EA–irMS) (bulk isotope analysis).
4.1.2. Molecular Sieving
Straight chain hydrocarbons are commonly separated from branched and cyclic hydrocarbons by treating saturated fractions with activated (250 °C, 8 h) 5A molecular sieve [62] in cyclohexane. The n-alkanes are recovered by dissolving the filter in HF (2 mL, 50% w/v) and neutralizing the solution with saturated sodium bicarbonate [9]. n-pentane is used to extract the aqueous phase (1 mL × 5). This fraction has n-alkanes that could be analyzed using gas chromatography–isotope ratio mass spectrometry (GC–irMS). Using a ZSM-5 molecular sieve, the branched and cyclic (5A excluded) hydrocarbon fractions from crude oils and sediments might be further simplified by removing methyl alkanes and alkyl cyclohexanes. The branched or cyclic (5A excluded) fraction is transferred to a small column (7–8 cm × 0.5 cm i.d.) of activated (250 °C) ZSM-5 molecular sieve (pre-rinsed with pentane) and allowed to stand (1–5 min). The branched or cyclic fraction is obtained after rinsing the filter with pentane (2 mL) (ZSM-5 excluded) [10].
4.1.3. Elemental Analysis–Isotope Ratio Mass Spectrometry (Bulk δ13C Analysis)
The principle behind determining the isotopic composition of individual molecules or the bulk stable isotope values of complicated mixtures is fairly similar. Regardless of the sample’s origin, each process necessitates complete combustion and conversion to CO2 and water [21]. For bulk determinations, the sample can be converted to CO2 using an elemental analyzer interfaced directly to an isotope ratio mass spectrometer in a sealed tube in the presence of CuO, or the sample can be converted to CO2 using an elemental analyzer interfaced directly to an isotope ratio mass spectrometer. A stable isotope ratio mass spectrometer is used to transfer and analyze CO2, which is made up of both 13CO2 and 12CO2. The relative amounts of 13CO2 and 12CO2 are measured in comparison to a standard material, Vienna Pee Dee belemnite (VPDB), which has a carbon isotope value of 0, given in the notation (see Table 1). Almost all samples will have lower 13C levels than the standard and will be depleted in 13C, resulting in negative 13C results. Stable carbon isotope ratios (R = 13C/12C) are expressed relative to the VPDB standard using the “delta” notation, where: δ 13C = (Rsample/Rstandard − 1) × 1000 (units are‰ or per mil or parts per thousand) [21]. The sample is weighed inside a small silver capsule, which is then folded and dropped into a pyrolysis reactor containing glassy carbon chips kept at 1260 °C for bulk D analysis. The sample is pyrolyzed to produce H2 and CO, as well as N2 if necessary. The pyrolysis products are separated on a 1 m chromatographic column packed with 5A molecular sieve and kept at 80 °C in an oven before being passed via a thermal conductivity detector (TCD) and into the irMS instrument. The D values are calculated and expressed in delta notation to VSMOW (Vienna standard mean ocean water) [10]. By precipitating barium sulfate, which is then combusted to produce sulfur dioxide, a custom-built stable isotope ratio mass spectrometer [63] can perform stable sulfur isotope ratio analysis. The sulfur isotopes are expressed in terms of the CDT standard. The ratios of the other elements, such as O, N, or Cl, are represented in the same way with their unique standard. Table 1 shows the natural abundance of the stable isotopes typically employed in geochemistry, as well as the international standards used to calculate their values.
4.1.4. Gas Chromatography–Isotope Ratio Mass Spectrometry (GC-IRMS)
A gas chromatograph coupled to the combustion system can allow the carbon isotope ratios to be determined on individual organic compounds (GC/combustion/IRMS, also called compound-specific isotope analysis (CSIA). After being eluted from the GC column, the components pass through a combustion reactor and are combusted to CO2 and water to determine the isotopic composition of individual compounds using GC-IRMS (Figure 1). The water is removed, and the CO2 is transferred to an isotope ratio mass spectrometer, which determines the relative amounts of 13CO2 and 12CO2 and calculates 13C values for each molecule. A Micromass IsoPrime isotope ratio mass spectrometer interfaced to a gas chromatograph for determining compound-specific stable carbon isotopic composition (13C values) or a chromatograph for determining stable hydrogen isotopic compositions (D values) is commonly used in GC–irMS [10]. For routine analysis, the oven is set to a temperature range of 40 to 310 °C at a rate of 3 °C/min, with initial and final hold times of 1 and 30 min, respectively. Samples are injected into an autosampler utilizing a split or splitless injector in pulsed splitless mode. Ultra-high purity (UHP) He is commonly utilized as carrier gas with a flow rate of 1.1 mL/min and a constant flow injector. The aromatic fractions of the samples are separated by a GC coupled to an inductively coupled plasma mass spectrometer to determine the sulfur isotope ratios of individual dibenzothiophenes. The operating conditions are described in detail in [64,65,66]. The samples are run in duplicates to ensure accuracy and the standard deviation between the two duplicates is noted accordingly [11]. The schematic diagram of GC-IRMS is shown in Figure 1 [21].
4.1.5. Methods of Stable Sulfur Isotope Analysis
SO2 is the gas of choice for gas–source mass spectrometric measurements. With the emergence of online combustion technologies [67], multistep offline preparations have been simplified to a single step, namely combustion in an elemental analyzer. Sample preparations have become less time-consuming and less reliant on potentially fractionating wet chemical extraction procedures, resulting in a minimum sample gas of less than 1 mg. Puchel et al. [68] and Rees [69] initially described a method that uses SF6 instead of SO2, which has several advantages, including no mass spectrometer memory effect and no need for raw data corrections of recorded isotope ratios because fluorine is monoisotopic. The reliability of the SO2 correction for oxygen isobaric interferences has been called into doubt by a comparison of 34S-values produced using the traditional SO2 and the laser SF6 techniques [70]. As a result, despite the high toxicity of the gases, the SF6 method has been revived [71], revealing that SF6 is an appropriate gas for detecting 33S/32S, 34S/32S, and 36S/32S ratios. Sulfur isotope ratios can now be determined using microanalytical techniques such as laser microprobe [71,72,73,74] and ion microprobe [75,76,77,78,79]. Hauri et al. [80] used NanoSIMS to collect data on several sulfur isotopes.
Bulk Stable Sulfur Isotope Analysis
Cai et al. [12] described the procedures for separating kerogen and analyzing sulfur isotopes in kerogen and oil. Pyrite is usually removed from kerogen by introducing a mixture of hot 6N HCl and CrCl2 to the ground dry kerogen under a nitrogen flow, with the H2S being carried to a trap and recovered as Ag2S. Water washing removes excess acids and acid-soluble ions from residual kerogen. The residual kerogen is collected and reground 2 h later to expose additional pyrite surfaces, and the process is repeated. Following the two treatments, the leftover kerogen is examined using X-ray diffraction (XRD) to see if pyrite levels are below detection limits (60.5% depending on conditions). If not, more treatments would be necessary. To oxidize organically linked sulfides to sulfate, a known weight (between 350 mg and 900 mg) of kerogen isolate or 1–4 g of oil is combusted in a Parr bomb apparatus at 25 atm oxygen. Total residual kerogen sulfur is calculated by precipitating dissolved sulfate as BaSO4 and weighing it. The maximum residual pyrite content in the kerogen after the chromium reduction is determined by measuring dissolved iron at pH < 2 with an atomic absorption spectrometer (assuming that all Fe occurs as pyrite in the kerogen). The produced BaSO4 is only evaluated for δ34S when leftover kerogen samples have contaminated pyrite sulfur and total sulfur < 0.08 to ensure low levels of errors (based on the differences in 34S value between kerogen and the related pyrite).
Individual Sulfur Compounds Isotope Analysis
Bendall et al. [81], Craddock et al. [82], and Parris et al. [83] have reported the use of multi-collector–inductively coupled plasma mass spectrometer (MC-ICP-MS) techniques. Amrani et al. [64] proposed a method for analyzing individual sulfur organic compounds using a gas chromatography–multi-collector–inductively coupled mass spectrometer (GC-MC-ICP-MS). Sample sizes are at orders of magnitude smaller than what is commonly used for SO2 and SF6 [84] due to poor detection limits. The GC-MC-ICP-MS method does not require any chemical preparation and allows for simultaneous collection of all four sulfur isotopes; however, it has lesser precision than other methods. The aromatic fractions of the oils and sediment extracts are separated by GC-MC-ICPMS to determine the sulfur isotope ratios of specific sulfur compounds. Amrani et al. [64,65] and Said-Ahmad and Amrani [66] described the system and its working conditions in detail. Duplicates for some of the oils and sediment extracts are measured and the standard deviation between the two duplicates is usually better than 1‰.
5. Applications of Stable Isotopes (C, H, S) in Shales and Crude Oils
Hydrogen isotopes are used occasionally in petroleum exploration to support biomarkers data but are widely used in gas exploration, while sulfur isotopes are rarely used in petroleum exploration. However, carbon isotopes are by far the most frequently used in gas and petroleum exploration. The following sections describe the uses of the isotopic compositions of carbon, hydrogen, and sulfur in petroleum and gas exploration.
5.1. Stable Carbon Isotope
The organic materials in a source rock must be characterized to determine if a formation will generate oil, gas, or both. Visual characterization, extraction, and identification of specific biomarkers and stable isotopes are all techniques that can be used to define organic matter [85]. Craig [86] and Silverman [2,87] discovered that oils from terrigenous sources were more depleted in 13C than oils from marine sources in the early studies of stable isotopes in hydrocarbon exploration. Land plants were generally deficient in 13C compared to aquatic plants and marine heterotrophs, which appeared to mirror the trend seen in living systems. Organic matter in recent sediments has also been found to become depleted in 13C as the depositional facies shifts from marine to terrigenous [88]. Carbon isotopes are frequently utilized to distinguish between oils originating from marine and non-marine organic matter. The relationships between the isotopic composition of the saturate and aromatic fractions [1] reveal differences between marine and non-marine oils. The effects of the source on stable carbon isotope ratios have been examined [89] and it was found that as the concentration of terrestrial input increases, the δ13C values become less negative. In west Texas, isotopic age trends were employed successfully to correlate and differentiate crude oils on a geographical basis [4]. Moreover, a general trend of lighter isotope enrichment with the increasing geological age of oils was observed, possibly caused by variations in photosynthesis intensity and changes in the isotopic composition of atmospheric CO2 [50,90]. This observation formed the basis for isotopic age-dating methods developed later [88]. By fractionating the oil into saturates, aromatics, NSO (nitrogen, sulfur, oxygen), and asphaltene fractions, the usage of bulk isotopes can be taken a step further to improve the precision. Individual fraction isotope data can then be plotted in a variety of ways. As shown in Figure 2, a plot of saturates vs. aromatics can be very useful for grouping oils formed from comparable source materials or distinguishing oils from distinct depositional settings [91].
The Stahl-type curves [92] can also be constructed using the isotope values. The stable isotope value for the kerogen can be integrated into this graph if the suspected source rock is available [21]. If the correct source rock is chosen, the individual fractions will have a direct link, as shown in Figure 3. The advantage of this method is that 13C values distinguish the carbon in different fractions and provide a more powerful correlation tool, though caution must be exercised in distinguishing the effects of increasing maturity [93], phase effects, and subsequent reservoir changes such as water washing [94] or biodegradation [95]. Understanding and quantifying these effects is therefore critical for the reliability of isotope correlation [96,97]. As a result, the factors that can alter the isotopic composition of crude oils and source rocks will be discussed in the following sections.
5.1.1. Source and Depositional Facies
Crude oil source rocks are generated from a variety of source materials that are deposited in a variety of depositional conditions [98]. Many factors will impact the isotopic compositions of these source materials, such as plants, phytoplankton, and bacteria, when they are first generated through photosynthesis. All of the original isotopic source signals will be homogenized and change slightly as this organic material is buried, degraded, and eventually heated to generate crude oils or natural gas [21]. However, because different crude oils are generated from diverse mixtures of source materials, some variations in the eventual isotopic composition of the producing crude oils are reasonable to expect. As a result, the isotopic composition of various crude oils from various sources is used.
As stated by the basic reaction below, the starting point in the generation of crude oil in atmospheric CO2 begins through the process of photosynthesis, which leads to the incorporation of that C into the cell wall components, providing energy to promote plant growth.
6CO2 + 12H2O ⟶ C6H12O6 + 6H2O + 6O2
The organic material from living plants and aquatic organisms is deposited in a variety of environments after they die. A substantial portion of this material will deteriorate depending on the environment, but a small part will be absorbed into the sedimentary record, go through diagenetic processes, and then thermally transform the residual organic matter to generate oil, gas, or both [21,98]. The level of fractionation between atmospheric 13CO2 and 12CO2 during photosynthesis varies depending on whether the source material is a land plant or a marine organism, and the extent of fractionation within land plants is further determined by distinct photosynthetic cycles.
The isotopic compositions of plants that were initially determined [86] showed that most of the plant materials investigated at the time had a rather stable δ13C value, about—27‰ in a thorough investigation on the carbon isotopic content of natural materials. It was noted that there did not appear to be any significant species or geographic effects; however, one grass with a δ13C value of 12‰ was later identified as a C4 plant. C3 plants, as defined by [86,99], incorporate CO2 from the atmosphere through carboxylation of ribulose bisphosphate. CO2 is absorbed by C4 plants by the carboxylation of phosphoenolpyruvate. The carboxylation product is transferred from photosynthetic cells’ outer layer (mesophyll cells) to the inner layer (the bundle sheath), where ribulose bisphosphate carboxylase decarboxylates and refixes it. C4 plants have lower negative δ13C values than C3 plants, according to isotope research. Plants with the C3 cycle, such as grasses, corn, and maize, grow in cooler, wetter climates and have isotope values in the 26 to 30 per mil range. Plants with the C4 cycle, such as grasses, corn, and maize, grow in dryer, hotter climates and have isotope values in the 10 to 14 range. The difference in the isotopic composition of C3 and C4 plants has become one of the primary methods for distinguishing these plant kinds [100], and it also serves as a useful diagnostic tool for distinguishing specific source materials.
Due to variations in the isotopic composition of carbon sources, numerous investigations of marine and terrestrial plants have demonstrated a depletion of 13C in land plants compared to marine plants [32,86]. Isotopic differences similar to this have been observed in recent sediments [13,53,54,101]. In comparison to local marine fauna and flora, organic matter in recent marine sediments is often depleted in 13C [102,103,104]. Diagenetic factors may be the cause of the 13C depletion. The 13C depletion has been attributed to the diagenetic loss of 13C-enriched carboxyl carbon [105,106]. Alternatively, it was proposed that the maturation of 13C-depleted lipids in marine sediments results in the formation of 13C-depleted organic matter [104]. However, selective preservation, in which 13C-depleted refractory components of algae are selectively accumulated in sediment and the 13C-enriched components are microbially degraded and lost, could explain the 13C depletion of marine sedimentary carbon [107]. Because source rocks are heterogeneous mixtures of many different types of organic material, the resulting isotopic composition of the source rock will be a weighted average of the isotopic compositions of all the different preserved and residual source materials, and thus the situation becomes far more complicated and less specific. As a result, depending on the initial mixture of source materials, there will be changes in the bulk isotopic compositions of the source rocks, and hence the oils. The significant contribution of terrestrial organic debris to the organic precursors of marine organic matter has been widely thought to explain the relative depletion in 13C of several marine sediment samples [2,5,108]. One explanation for differences in the resulting crude oils is isotopic variations in the source materials.
Moreover, there exists a comprehensive geochemical analysis of oils from offshore Brazil, using a variety of characteristics to distinguish oils from distinct depositional settings [109]. Based on a variety of criteria, including δ13C values, the oils were categorized into five groups (I to V). Table 2 summarizes some of the bulk parameters for each category of oils, including bulk isotope analyses, elemental analyses, and alkane distributions. Figure 4 shows how the oils were grouped into five types using δ13C values of the saturated and aromatic hydrocarbons. Group II oils are isotopically heavy (δ13C values about 25‰), which could be explained by increased salinity. Carbonate complexes are preferred as a carbon source for photosynthesis by plants from saline environments. These have a higher 13C content than atmospheric carbon dioxide, which has a higher 12C content [110].
5.1.2. Biodegradation
Biodegradation has a well-defined set of changes that occur during the degradation process [85]. The primary changes in isotopic composition, on the other hand, are due to bulk changes in the oil rather than changes in individual molecules. The biodegradation of crude oil in the reservoir is a significant secondary alteration process with significant economic implications. Although the actual mechanisms involved in the in-reservoir biodegradation of crude oil are still being studied (e.g., site and rate of degradation, nutrient availability, and nature of by-products), their impact on the composition and physical qualities is well documented [111]. In most cases, the components of crude oils are removed sequentially in the order of n-alkanes > monocyclic alkanes > alkyl benzenes > isoprenoid alkanes > alkyl naphthalenes > bicyclic alkanes > steranes > hopanes. Many compound-related markers for determining the level of crude oil biodegradation have been proposed based on this sequence [112]. Biodegradation’s effects on the isotopic compositions of individual compounds have been explored for prospective use in petroleum and environmental research [57,113,114,115,116,117,118,119,120,121]. The effects of slight and moderate biodegradation on light hydrocarbons (C5–C9) consistently lead to 13C enrichment for each surviving molecule, according to studies by [57,119]. However, slight and moderate biodegradation resulted in insignificant enrichment in residual C10–C14 n-alkanes, with a maximum δ13C increase of 0.5‰, according to [111,114]. The stable carbon isotope compositions of natural gas components (C2–C5) are significantly fractionated during biodegradation, as has long been observed [122]. During biodegradation, lower carbon number molecules are removed first, followed by higher carbon number ones. Except in extreme cases of biodegradation, the amount of change in the aromatic and polar fractions is negligible [1,13]. The impact on the isotopic composition of the saturate fraction, on the other hand, can be much greater because n-alkanes make up a large portion of the saturate fraction, and their removal alters the isotopic composition, causing the saturate fraction to become isotopically heavier in general, as shown in the Stahl diagram in Figure 3.
5.1.3. Maturity
Unlike biodegradation, maturity can have an impact on the isotopic composition of all fractions, as maturity causes carbon–carbon bonds to break in all fractions, resulting in increased proportions of lighter hydrocarbons. Crude oil is formed by burial and maturity, and as a result of thermal maturation, there will be some variation in isotope composition. The effects of maturation on the 13C values of kerogens in the natural system have been thoroughly studied [123,124]. The kinetic isotope effect dominates during the maturation phase, resulting in the preferential breaking of bonds containing the lighter isotope. As a result, the heavier isotope will be enhanced in the residual fractions, and the overall trend will be one of isotopic enrichment as maturity increases. The degree of enrichment will differ from one oil to the next and will also be determined by the level of maturation. Oil-to-gas cracking degrades oil by turning it into low molecular weight molecules in a reservoir. Isotopic effects are more likely to represent kinetic isotope fractionation in this scenario, as 12C–12C bonds are preferentially cleaved over 13C–12C bonds, leaving the residual oil enriched in 13C [96,97]. The residual kerogen will become isotopically heavier with increasing maturity of source rocks, and the cracking of the kerogen within the source rock to generate oil will have similar effects. The products formed from kerogen will initially be isotopically light, but as maturity levels rise and more isotopically enriched kerogen is thermally degraded, the resulting products will become isotopically heavier. In general, kinetic isotopic fractionation in processes involving compounds with six or more carbon atoms is insignificant [96,97]. An additional isotopic fractionation step must be considered for condensates or light oils. As the subsurface gas phase (“gas condensate”) is raised to the surface and temperature and pressure decrease, condensates (usually C6+ compounds) condense from the subsurface gas phase (“gas condensate”). Because we are dealing with a transition from a liquid state (“oil”) to a vapor state (“condensate”), isotope effects will be limited during this process. Any isotope effect will be due to equilibrium isotope effects rather than the kinetic isotope effect in this case. However, in comparison to the oil from which they emerged, condensates will preferentially accumulate smaller molecular weight molecules. Differences in bulk δ13C between low and high molecular weight molecules will be reflected in bulk δ13C values for oil and condensate [96,97]. There are two ways to look into isotope effects that occur during the maturation and generation process. First, accumulated oils derived from a single source rock but maturing at distinct rates based on biomarker maturity characteristics can be isotopically identified, with discrepancies in the δ13C values attributed to maturation. This also presupposes that no further in-reservoir changes have happened. Laboratory maturation investigations, such as hydrous pyrolysis experiments [125], where an immature source rock sample is heated in the presence of liquid water at subcritical temperatures (<374 °C), provide an alternative option. Although these tests use relatively high temperatures (300–365 °C) to compensate for the long period required to generate oil in the natural system (106–109 years), the pyrolysates released from potential source rocks are similar to natural crude oil [125]. The vitrinite reflectance and atomic H/to C ratio of pyrolysates, which were measured in the matured source rocks, revealed similar alterations to those found in the natural system [124,126]. Immature source rock samples from the Woodford Shale were artificially matured to see how thermal maturation affected the δ13C values of kerogen, bitumen, and crude oil. It was discovered that as thermal maturation increased, the expelled oil became more enriched in 13C. (Figure 5). As stated above, the increased cleavage of 13C–12C bands in the bitumen as it continues to decompose into expelled oil and preferential cleavage of 12C–12C bonds in the expelled pyrolysate as it decomposes into gases and pyrobitumen can be attributed to the kinetic isotope effect and increased cleavage of 13C–12C bands in the bitumen as it continues to decompose [21].
5.1.4. Age Dating
To age date oil samples, exploration studies have used several biomarker parameters that can be linked to certain evolutionary processes [127]. The interest in age dating stems from the fact that in many exploratory cases, only oil samples are recovered, and being able to anticipate the age of the source rock from crude oil properties is extremely important in determining the source formation responsible for the development of a certain oil. Andrusevich et al. [88] studied the possibility of age dating crude oils using stable carbon isotope compositions of different fractions of oil samples and found that the oils became more enriched in 13C as geologic age decreased (Figure 6). Changes in δ13C, in particular, are linked to global fluctuations in 13C carbonate carbon [88]. Previous studies of the stable carbon isotopic composition of oils (δ13C) over geologic time have found a general tendency of 13C enrichment with decreasing age [20,128]. The δ13C value of oil is often determined by the δ13C value of the kerogen in the source rock from which it was derived. The δ13C value of kerogen is determined by the sorts of organisms maintained as well as the δ13C values of its substrate, which leads back to the original photosynthetic conditions as stated above. Assuming that the present is the key to the past, variables known to be essential in the modern biosphere, such as temperature, pCO2, and depositional settings, are likely to have influenced the stable isotope composition of carbon in ancient biospheres. Organic carbon and carbonate carbon production, as well as their corresponding δ13C values, would have been influenced by changes in these settings [129]. The geologic record includes changes in the relative quantities of atmospheric gases such as CO2 and O2 as well as fluctuations in the δ13C values of organic and carbonate carbon [21,129]. Variations in carbon isotope values over time can be explained very readily. The first CO2 in the atmosphere came from the outgassing of the core and had a fixed isotopic composition [21,128]. The CO2 content of the atmosphere declined over time as more complex photosynthetic creatures and plants evolved, and the atmosphere became isotopically heavier in general [20,21,128]. There were isotopic excursions where CO2 became even heavier as a result of considerable productivity increases, and the principal photosynthesizers took on a heavier isotopic signature as a result. Because most fossil fuels and their combustion products have isotopic values in the range of 25 to 35 per mil [21], the isotopic composition of atmospheric CO2 is becoming lighter as the burning of fossil fuels increases.
5.1.5. Carbon CSIA
It took over two decades to develop the ability to determine the isotopic composition of individual compounds. Continuous flow determination of carbon isotope values of individual compounds in complex mixtures of geochemical interest is possible with gas chromatography–isotope ratio mass spectrometry (GC–IRMS) [130,131,132,133]. Only bulk isotopic data were available before the introduction of GC–IRMS [102], which lack a lot of specificity due to the heterogeneous character of the source material [134]. As a result, isotopic data from crude oils and source rock extracts must be interpreted in conjunction with other geochemical information [135]. The use of GC-IRMS now allows the isotopic composition of individual compounds in crude oils and source rock extracts to be determined. The n-alkanes and isoprenoids have received the greatest attention since they are often well resolved in chromatograms [136]. Individual n-alkane δ13C values have been widely employed in oil–oil and oil–source rock correlations [137,138,139] as well as reconstructions of the paleoenvironment and paleoclimate [132,140]. The isotopes of individual compounds provide an additional correlation parameter, as well as the ability to determine whether or not many compounds are derived from the same precursor molecule. This is especially true for the pristane to phytane ratio [141], which is one of the most extensively used geochemical parameters in geochemical investigations. This ratio is commonly utilized for correlation as well as indicating the depositional environments and redox conditions. However, this latter application assumes that both compounds are derived from the side chain of chlorophyll, and if they are not, the ratio is invalid. The only way to determine whether they are from the same source is by measuring the carbon isotope value of these substances. A carbon isotopic investigation of crude oils and the Messel shale was conducted [131], which demonstrated that the isotopic values in the oils were quite comparable, indicating that they came from the same source, but the values in the Messel extract were significantly different, indicating that the pristane and phytane came from distinct origins. Isotopic compositions of individual compounds have also been successfully used to characterize specific components in the saturated hydrocarbon fraction and as a parameter to establish oil–oil and oil–source rock correlations [53,54,137,142,143]. This technique has been used by several researchers to assess the presence of uncommon polycyclic alkanes in extracts of source rocks and crude oils where hopanoids and steranes were either absent or present in extremely low quantity [144,145,146,147,148]. The assumption behind the oil–source rock correlation is that the oil and the corresponding source rock have similar δ13C values. To use isotopic compositions from crude oils and source rock extracts for direct oil–oil and oil–source correlations, two pre-requisites must be met: first, the maturity level of the oil and corresponding source rock must be similar; and second, secondary alterations such as hydrocarbon expulsion, migration, biodegradation, and water washing, among others, must have had no significant effect on the isotopic composition of either the oil or potential source rock [149,150]. Regardless of whether bulk or compound-specific isotopes are being identified, these requirements must be met. Carbon isotope profiles of n-alkanes can easily distinguish source rock samples from different areas within a basin, as demonstrated by [151], who analyzed δ13C values of specific n-alkanes for different formations throughout the Liaohe Basin in China. The n-alkanes of one of the Es4 members have a relatively constant isotopic profile, with δ13C values ranging from 28 to 32‰, whereas the n-alkane δ13C values for the other Es3 source rocks range from 23 to 28‰, with a trend toward isotopically lighter values with increasing n-alkane chain length, typically 2–5‰ [152,153]. The n-alkanes found in the samples have similar characteristics to the Es4 source rocks, implying a saline lacustrine environment that is rather closed (Figure 7; [151]).
5.1.6. Application of Stable Carbon Isotopes to Gas Exploration
Natural gases, unlike crude oils, are generally simple mixtures, limiting the number of factors that can be used to differentiate gases from different sources [21]. Hydrocarbons in the C1 to C5 range will predominate in most natural gas samples, with varying quantities of CO2, H2S, and N2 [21]. The maturity of the source rock or oil from which the gas was formed will, to a significant extent, affect the proportions of individual components. At high levels of thermal maturity, methane-dominated gases are generated, whereas at low levels of thermal maturity, biogenic activity can result in methane-dominated gases. Differentiating biogenic from thermogenic methane is one of the more common uses of stable isotopes. Biogenic gas has a very light isotope, often around 70 per mil, whereas thermogenic methane has an isotope of 30 to 50 per mil [21,154]. This was realized many years ago, which led to one of the first correlation or source discrimination diagrams [154]. Essentially, this is a cross plot of methane δ13C vs. C1/C2+ ratio, and as seen below, biogenic samples are characterized by isotopically light methane and a high C1 to C2+ ratio (Figure 8). Thermogenic gas samples will be heavier isotopically and have lower compositional ratio values. There is also a mixed gas area were assigning a specific source to the gases in that region is a little more challenging. The kinetic isotope effect is responsible for the variance in these isotopic ratios [21,154]. Bacteria preferentially cleave 12C–12C bonds, resulting in the formation of isotopically light biogenic gas. From a thermogenic standpoint, these bonds take less energy to break at low levels of maturity than a 12C–13C bond. As the maturity level rises, more bonds containing the heavier isotope will be broken, causing the methane produced to become heavier isotopically. Natural gas samples have fewer alternatives for correlation or discrimination than crude oil samples, as previously stated [21,154]. The isotopic composition of the individual compounds in the gas samples, as well as variations in the relative quantities of the individual compounds, must be used to make these associations [21,155]. The GC-IRMS technique has been shown to be quite beneficial, making it comparatively simple to determine the isotopic compositions of individual compounds in complex mixtures. Before the invention of the GC-IRMS, each compound had to be physically isolated to determine its isotopic composition.
With the GC-IRMS system, it is of course possible to simply introduce the sample into the GC and obtain the isotopic composition of each component in a relatively short period. It is also worth noting that these individual compounds can have both carbon and hydrogen isotope values, and a combination of the two isotopes offers an extra correlation parameter [21,154,155]. Simply plotting the isotope levels of specific compounds against their carbon number is the most useful application of isotopes for correlation purposes. Then, with a sufficient number of gas samples from the same basin, those that are connected will plot together, but those from other sources or created at different levels of maturity will plot independently, as illustrated in Figure 9 [21].
This graph has many interesting extensions that can reveal a significant amount of additional information. As previously stated, we are constrained in our ability to distinguish between natural gas sources. However, after examining the isotopic composition of the individual compounds in the condensates, it has been proven that for wells that produce both gas and condensates, the plots presented above can be extended into the condensate range. The isotope values for the individual compound in the condensate will simply be an extension of the relevant gas sample, as illustrated if the condensate and gas are generated from the same source and at the same maturity level (Figure 10) [155]. The benefit of collecting these extended plots is that we can utilize the isotopes to establish the relationship between gas and condensate, but there are also biomarkers, such as diamondoids, in the condensates that may be used to correlate or discriminate between samples [21,155].
5.2. Stable Hydrogen Isotope
Hydrogen isotope has been applied to oil–oil and oil–source rock correlations in petroleum geochemistry [18,156]. The δD signature of the precursor must be conserved throughout sedimentation, burial, diagenesis, and catagenesis for the stable hydrogen isotopic composition (δD) of sedimentary hydrocarbons to resemble that of their biosynthetic precursors [10]. Over long periods of geological time (millions of years), diagenetic and catagenetic effects are thought to induce considerable hydrogen (H/D) exchange between organic hydrogen and hydrogen species in the surrounding environment [9,14,19,157,158,159,160,161,162,163]. Thermal maturation, in particular, has been discovered to play an important role in the change of indigenous δD signatures, with increasing maturity resulting in a general enrichment of D in hydrocarbon fractions and individual hydrocarbons [14,19,162,163]. The hydrocarbons are expected to be exchanging hydrogen with the comparatively D-rich formation fluids [158,164]. The influence of maturation on the δD values of sedimentary hydrocarbons (n-alkanes, pristane, and phytane) in a sequence of marine source rocks from the Perth Basin was demonstrated by [162] (Western Australia). Immature source rocks (%R0 = 0.53) have distinct δD signatures, with pristane and phytane considerably reduced in deuterium (D) compared to the n-alkanes. The discrepancy between the δD values of n-alkanes and isoprenoids gradually reduces as maturity increases (up to %R0 = 1.13). Pristane and phytane become increasingly D-enriched, while then-alkanes maintain a steady isotopic composition until late maturity when D-enrichment in n-alkanes becomes considerable. Isotopic exchange processes linked with thermal maturation have been attributed to the D enrichment.
A similar trend of D enrichment in n-alkanes and isoprenoids was observed in other studies [14,163]. The former study presented the effects of thermal maturity on n-alkane and isoprenoids δD levels in two sediment sections in Poland (Kupferschiefer, KS) and Germany (Posidonia Shale). With increasing age, all n-alkanes and isoprenoids were enriched in D, with isoprenoids enriching at a faster rate than n-alkanes. The latter investigated a 450-meter core of Early Cretaceous lacustrine sediments from West Africa, which ranged in maturity from immature to early mature (%R0 0.55–0.7). The effects of maturation on the δD values of individual sedimentary hydrocarbons from sedimentary sequences with a wider maturity range (%R0 0.6–1.6), such as Paqualin-1 and Vulcan-1B from the Vulcan Sub-basin offshore northern Australia has been analyzed [10]. The enrichment in D in isoprenoids was found to be connected to the epimerization of pristane and phytane and to correspond substantially with traditional maturity criteria. D enrichment was seen in pristane and phytane isolated from post-mature Paqualin-1 sediment, demonstrating that D enrichment was maintained at extremely high maturity, more so for regular isoprenoids than n-alkanes. This corroborated the theory that hydrogen (H/D) exchange, rather than initiating free radical hydrogen transfer, causes the observed shift in δD values. In the sedimentary environment, a mechanism was postulated to account for both H/D exchange and the epimerization of pristane and phytane. Pristane was found to be more enriched in D than phytane across the Vulcan Sub-basin sequences, indicating that they exchanged hydrogen at equal rates during maturation. This was attributed to a reduced relative algal input to the isoprenoids [10].
5.2.1. Hydrogen CSIA
δD measurements of bulk OM or whole fractions from crude oils or sediment extracts were the only way to analyze the distribution of stable hydrogen isotopes in sedimentary OM. Because aliphatic C-bound hydrogen is probably the most isotopically conservative, isotopic analysis of individual aliphatic compounds with solely C-bound hydrogen (e.g., n-alkanes) is appealing. Compound-specific hydrogen isotope analysis [18,165] has made it possible to determine the D/H composition of individual compounds in complex mixtures. Because it has the highest mass difference between its two stable isotopes (D and H), and hence the greatest natural variability in stable isotope ratios of all elements, hydrogen compound-specific isotope analysis has shown remarkable promise in petroleum geochemistry. The existing literature has looked into the relationship between δD values of whole crude oils and bitumen sand and their origins (including organic matter type and depositional circumstances), thermal maturity, and secondary processes such as biodegradation [147], mixing [10], and migration [147]. In a different study, it was explained how different organisms fractionate hydrogen isotopes in the production of lipids [166] while another one reported individual n-alkanes and isoprenoids δD values as evidence of large and rapid climate change [9]. Furthermore, the efficacy of hydrogen CSIA in petroleum correlation studies using a large number of crude oil samples from the Western Canada Sedimentary Basin was illustrated [10]. Additionally, it was shown that sedimentary aliphatic hydrocarbon D values can be utilized to determine the age of source rocks and crude oils from the Perth Basin (WA) and Vulcan Sub-basin (Timor Sea) [9,147,166,167]. Pristane (Pr) and phytane (Ph) have substantially lower δD values than n-alkanes; however, this difference diminishes with age due to thermal hydrogen isotope exchange. These studies suggested that the δD measurements of sedimentary hydrocarbons are a good source of the effects-aware maturity metric. For Devonian source rocks from the Western Canada Sedimentary Basin (WCSB), the D/H of biomarkers has been employed as a maturity proxy [147]. Additionally, how various organisms fractionate hydrogen isotopes in lipid production was investigated [168]. The δD values of compound-specific lipid biomarkers extracted from peat deposits [15] and sediments [16] have been utilized as a proxy for palaeoenvironmental and palaeoclimatic conditions. During the Messinian salinity crisis, δD values of individual n-alkanes and isoprenoids were reported as evidence of substantial and rapid climatic fluctuation [147]. Using crude oil samples from the Western Canada Sedimentary Basin, the utility of hydrogen CSIA in petroleum correlation studies and palaeoenvironmental reconstructions was evaluated [169]. Next, the ability of lipid δD values to be preserved in Miocene lacustrine sediments and plant fossils from Clarkia, Idaho, USA was demonstrated [170]. The Clarkia sediments they looked at are the oldest samples known (15–20 million years) with original δD values that appear to have been retained. There is concern that long-term diagenetic effects can result in considerable hydrogen isotope exchange between organic hydrogen and the surrounding environment [157,158,159,160,161,171]. Thus, the extent to which hydrogen CSIA can be employed for palaeoenvironmental investigations, particularly when applied to older sedimentary organic matter, is of interest. Organic geochemists and palaeoclimatologists are interested in the δD values of organic compounds preserved in sediments because they can represent the isotopic composition of water in ancient environments. Because water mobility and the energy transferred as it changes physical states are major elements in weather and climate, hydrogen isotopic fractionations are assumed to be linked to a range of naturally occurring events in the hydrological cycle. ‘Meteoric’ waters are those that have gone through the hydrological cycle. Temperature, altitude, latitude, closeness to the ocean, and other factors affect the isotopic values of meteoric waters [50,172]. Water is the primary hydrogen source for photosynthetic organisms, and the deuterium concentration in the source water is mirrored in the deuterium composition of the organism [168]. The remains of species are eventually absorbed into sediments and added to petroleum’s organic matter. Similarly, CSIA of hydrogen to investigate Late Carboniferous to Late Permian torbanites from Torbane Hill, Scotland, and the southern hemisphere, eastern Australia, was used [17], where it was discovered that the δD values of n-alkanes and isoprenoids were similar to those found in modern biological samples, which implies that their indigenous δD signatures may have been preserved for at least 260–280 million years.
5.2.2. Application of Hydrogen Isotope to Gas Exploration
The hydrogen isotopes of alkane gases, as well as other saturated and aromatic hydrocarbons combined with carbon isotopes, play a critical role in the identification of natural gas genetic-type analysis, parent material source, maturity, mixing, biodegradation, and (TSR) [147,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188]. Apart from the parent material type, maturity, biodegradation, and TSR, the water environment during the deposition and diagenesis of source rock (such as salinity) also plays a crucial effect [147,189]. Many studies have been conducted on the hydrogen isotopic characteristics of natural gases from the Ordos Basin Permian and the Upper Triassic Xujiahe Formation of the Sichuan Basin; the origin and source of natural gases have also been analyzed based on the carbon and hydrogen isotopic and chemical composition characteristics of the alkane gas. The natural gases in the above two places have been proven to be coal-formed gas [190,191,192,193,194,195,196,197,198,199,200], and hydrogen isotopic indices for detecting natural gas genesis and vitrinite reflectance (Ro) values have been proposed [147,197]. Some studies on factors affecting hydrogen isotopes of alkane gas have been conducted [167,197], but most of them focused on the composition of hydrogen isotopes and affecting factors of Permian natural gases (methane and its homologs) and the Upper Triassic Xujiahe Formation in the two basins from a single basin perspective, and a few made comparisons [197], but only for methane hydrogen isotopes. The composition, influencing factors, maturity, and natural gas genetic identity indicators of hydrogen isotopes of heavy alkane gases, such as ethane and propane, received little attention. In a recent study [201] the hydrogen isotopic compositions of coal-formed Permian gas in the Ordos Basin and Upper Triassic gas in the Sichuan Basin, and the hydrogen isotopic compositions of alkane gas in various areas were also studied. To develop and improve the coal-formed gas theory and genetic identification theory of natural gas, as well as to guide natural gas exploration, the influencing factors and their degree of influence on the hydrogen isotopic composition of alkane gases were investigated, and hydrogen isotope indexes of alkane gas capable of identifying the natural gas genetic type and Ro value of gas were proposed.
5.3. Stable Sulfur Isotopes
Sulfur isotopes have been utilized to correlate source rocks and oils in a rapidly buried basin that have not been altered by thermochemical sulfate reduction (TSR) [11,12,202,203,204]. Hydrocarbons are produced quickly in such a basin, and peak oil is anticipated to occur under semi-closed to closed circumstances. In case studies and experimental simulations, this property, together with strong H2S solubility and quick sulfur isotope homogenization, is thought to result in modest discrepancies (up to 2) in 34S values between mature kerogens and their generated oils [12]. With the δ34S values reported in closed range in both the oils and the source rocks, sulfur isotopes have been effectively used to correlate Cambrian-derived oils with Cambrian source rocks [11,12]. To identify the source of oils and oil-source rock correlations in oils and source rocks from the Tazhong area, Tarim Basin, China, bulk and individual n-alkane δ13C and individual alkyldibenzothiophene δ34S values were used [205]. The majority of the oils from the Tazhong area were most likely generated from Cambrian source rocks, and δ13C and δ34S values can be utilized to indicate the oil–source rock relationship in the Tazhong uplift, Tarim Basin, China. Sulfur isotopes have also been successfully used for oil–oil correlations in the oils from the Williston Basin of North Dakota and Saskatchewan [202]. The sulfur isotopic composition can be utilized to reconstruct changes in the sedimentary environment since large cycles between sulfate and sulfide are associated with organic and inorganic carbon and iron cycles [205,206,207]. A series of geological and geochemical studies, including a pyrite sulfur isotopic investigation of lacustrine black shales from the Yanchang Formation in Ordos Basin, China was recently carried out [208]. The paleoredox chemical conditions were confirmed to be the trigger for the anomalous sulfur isotopic compositions based on the analysis of sulfur isotope fractionation during the sulfur cycle.
6. Conclusions
This paper critically reviewed the early and recent trends in stable isotope geochemistry of organic elements in shales and crude oils. The bulk and compound-specific stable isotopes of H, C, and S, as well as their uses as source facies, depositional environments, biodegradation, thermal maturity, geological age, and oil–oil and oil–source rock correlation studies, were discussed. The applications of the stable isotopes of H and C to gas exploration were also discussed. Then, the experimental and instrumental approaches in the stable isotopes of H, C, and S were discussed. This review showed that the stable isotopes of H, C, and S could be utilized to establish the origin, depositional conditions, and thermal maturity of organic matter of shales, as well as oil–oil and oil–source rock correlation studies, which are critical to hydrocarbon exploration. However, there is limited literature that discussed the sulfur isotopic compositions of individual biomarkers in crude oils, while papers on compound-specific isotopic analysis of individual biomarkers in shales are very rare in the literature. The reason is that these compounds are present in relatively low concentrations and generally cannot be seen on a regular gas chromatogram; hence, it is not possible to obtain isotopic compositions of individual biomarkers on a routine basis, especially sulfur. The need here is to develop more routine molecular sieving methods that can be used to isolate concentrates of the biomarkers and then determine the isotopic composition of these compounds and provide another fingerprinting tool for correlation purposes. Conclusively, emerging isotopic techniques such as laser microprobe, ion microprobe, and NanoSIMs, which can collect data on the surface, are recommended for further stable isotopic studies on shales and crude oils.
Author Contributions
Conceptualization, A.B.O., B.L. and M.O.; writing—original draft preparation, A.B.O.; writing—review and editing, A.B.O., B.L. and M.O.; supervision, B.L.; project administration, B.L.; funding acquisition, B.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Science and Technology Project of Heilongjiang Province (2020ZX05A01).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors appreciate the anonymous reviewers and editors, whose constructive comments have greatly improved the quality of this paper.
Conflicts of Interest
No conflicts of interest among the authors.
References
- Sofer, Z. Stable Isotopes in Petroleum Exploration Source and Migration Process and Evaluation Techniques; Amoco Production: Tulsa, OK, USA, 1991; pp. 103–106. [Google Scholar]
- Silverman, S.R.; Epstein, S. Carbon isotopic composition of petroleums and other sedimentary organic materials. Am. Assoc. Pet. Geol. Bull. 1958, 42, 998–1012. [Google Scholar]
- Silverman, S.R. Investigations of Petroleum Origin and Evolution Mechanisms by Carbon Isotope Studies. In Isotopic and Cosmic Chemistry; Craid, H., Miller, S.L., Wesserburg, G.J., Eds.; North-Holland Publishing Company: North-Holland, Amsterdam, 1964; pp. 92–102. [Google Scholar]
- Kvenvolden, K.A.; Squires, R.M. Carbon isotopic composition of crude oils from Ellenburger group (Lower Ordovician), Permian basin West Texas and Eastern New Mexico. Am. Assoc. Pet. Geol. Bull. 1967, 5, 1293–1303. [Google Scholar]
- Eckelman, W.R.; Broecker, W.S.; Whitlock, D.W.; Allsup, J.R. Implications of carbon isotopic composition of total organic carbon of some recent sediments and ancient oils. Am. Assoc. Pet. Geol. Bull. 1962, 46, 699–704. [Google Scholar]
- Alexeyev, F.A.; Lebedev, V.S.; Krylova, T.A.; Ovsyannikov, V.M.; Grachev, A.V. Carbon Isotopic Composition of Natural Hydrocarbons and the Problems of Their Origin; ONTI VNIYaGG: Moscow, Russia, 1967. [Google Scholar]
- Anderson, T.F.; Pratt, L.M. Isotopic Evidence for the Origin of Organic Sulfur and Elemental Sulfur in Marine Sediments. ACS Symp. Ser. 1995, 612, 378–396. [Google Scholar]
- Habicht, K.S.; Canfield, D.E. Isotope fractionation by sulfate-reducing natural populations and the isotopic composition of sulfide in marine sediments. Geology 2001, 29, 555–558. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R. Effect of maturation on the indigenous delta D signatures of individual hydrocarbons in sediments and crude oils from the Perth Basin (Western Australia). Org. Geochem. 2005, 36, 95–104. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R.; Edwards, D. The effect of source and maturity on the stable isotopic compositions of individual hydrocarbons in sediments and crude oils from the Vulcan Sub-basin, Timor Sea, Northern Australia. Org. Geochem. 2007, 38, 1015–1038. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, C.; Worden, R.H.; Wang, T.; Li, H.; Jiang, L.; Huang, S.; Zhang, B. Application of sulfur and carbon isotopes to oil–source rock correlation: A case study from the Tazhong area, Tarim Basin, China. Org. Geochem. 2015, 83–84, 140–152. [Google Scholar] [CrossRef]
- Cai, C.; Li, K.; Anlai, M.; Zhang, C.; Xu, Z.; Worden, R.H.; Wu, G.; Zhang, B.; Chen, L. Distinguishing Cambrian from Upper Ordovician source rocks: Evidence from sulfur isotopes and biomarkers in the Tarim Basin. Org. Geochem. 2009, 40, 755–768. [Google Scholar] [CrossRef]
- Sofer, Z. Stable Carbon Isotope Compositions of Crude Oils: Application to Source Depositional Environments and Petroleum Alteration. AAPG Bull. 1984, 68, 31–49. [Google Scholar]
- Radke, J.; Bechtel, A.; Gaupp, R.; Pu¨ttmann, W.; Schwark, L.; Sachse, D.; Gleixner, G. Correlation between hydrogen isotope ratios of lipid biomarkers and sediment maturity. Geochim. Cosmochim. Acta 2005, 69, 5517–5530. [Google Scholar] [CrossRef]
- Xie, S.; Nott, C.; Avsejs, L.; Volders, F.; Maddy, D.; Chambers, F.; Gledhill, A.; Carter, J.; Evershed, R. Palaeoclimate records in compound-specific δD values of a lipid biomarker in ombrotrophic peat. Org. Geochem. 2000, 31, 1053–1057. [Google Scholar] [CrossRef]
- Sauer, P.; Eglinton, T.; Hayes, J.M.; Schimmelmann, A.; Sessions, A. Compound-specific D/H ratios of lipid biomarkers from sediments as a proxy for environmental and climatic conditions. Geochim. Cosmochim. Acta 2001, 65, 213–222. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Wang, S.X.; Alexander, R.; Radke, J. Stable hydrogen isotopic composition of hydrocarbons in torbanites (Late Carboniferous to Late Permian) deposited under various climatic conditions. Org. Geochem. 2004, 35, 189–197. [Google Scholar] [CrossRef]
- Asif, M.; Fazeelat, T.; Grice, K. Petroleum geochemistry of the Potwar Basin, Pakistan: 1-Oil–oil correlation from biomarkers and δ13C/δD. Org. Geochem. 2011, 42, 1226–1240. [Google Scholar] [CrossRef]
- Rigby, D.; Batts, B.; Smith, J. The effect of maturation on the isotopic composition of fossil fuels. Org. Geochem. 1981, 3, 29–36. [Google Scholar] [CrossRef]
- Hoefs, J. Stable Isotope Geochemistry, 8th ed.Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Philp, R.P.; Monaco, G.L. Applications of Stable Isotopes in Hydrocarbon Exploration and Environmental Forensics. In Advances in Isotope Geochemistry; Mark, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 639–677. [Google Scholar]
- Tissot, B.T.; Welte, D.H. Petroleum Formation and Occurrences: A New Approach to Oil and Gas Exploration; Springer: Berlin/Heidelberg, Germany, 1978. [Google Scholar]
- Frank, D.; Sackett, W. Kinetic isotope effects in the thermal cracking of neopentane. Geochim. Cosmochim. Acta 1969, 33, 811–820. [Google Scholar] [CrossRef]
- Park, R.; Dunning, H. Stable carbon isotope studies of crude oils and their porphyrin aggregates. Geochim. Cosmochim. Acta 1961, 22, 99–105. [Google Scholar] [CrossRef]
- Muller, P.; Wienholz, R. Bestimmung der natürlichen Variationen der Kohlenstoffisotope in Erdöl-und Erdgaskomponenten und ihre Beziehung zur Genese. Z. Angew. Geol. 1967, 13, 427–450. [Google Scholar]
- May, F.; Freund, W.; Mu¨ller, P. Modellversuche über Isotopenfraktionierung von Erdgaskomponenten während der Migration. Z. Angew. Geol. 1968, 14, 376. [Google Scholar]
- Colombo, U.; Gazzarrini, F.; Sironi, G.; Gonfiantini, R.; Tongiorgi, E. Carbon isotope composition of individual hydrocarbons from italian natural gases. Nature 1965, 205, 1303–1304. [Google Scholar] [CrossRef]
- Welte, D.H. Der 13C-Isotopengehalt von geradzahligen und ungerazahligen höheren n-parafinen Paraffinen aus Erdöl. Erdöl und Kohle 1969, 22, 150–162. [Google Scholar]
- Botneva, T.A.; Müller, P.; Maas, I. On carbon isotopic composition of oils and their fractions. Geol. Nefti Gaza 1969, 7, 33–39. [Google Scholar]
- Lebedev, V.S. Isotope composition of oil and gas. Geokhimiya 1964, 11, 1128–1137. [Google Scholar]
- Galimov, E.M. 13C enrichment of methane during passage through the rocks. Geochem. Int. 1967, 4, 1180–1181. [Google Scholar]
- Galimov, E.M. Geochemistry of Carbon Stable Isotopes; Nedra: Moscow, Russia, 1968; p. 224. [Google Scholar]
- Galimov, E. Isotopic composition of carbon in gases of the crust. Int. Geol. Rev. 1969, 11, 1092–1104. [Google Scholar] [CrossRef]
- Galimov, E.M. Die Isotopenzusammensetzung des Koh-lenstoffs in den Gasen der Erdkruste. Z. Angew. Geol. 1969, 15, 69–81. [Google Scholar]
- Galimov, E.M. On the relationship of the fractionation coefficient of isotopes to the equilibrium constants of the isotope exchange reactions of carbon in hydrocarbon systems. Zh. Fiz. Khim. 1971, 45, 1187–1191. [Google Scholar]
- Galimov, E.M.; Kuznetsova, N.G.; P’yankov, N.A.; Vinnikov-ski, S.A. Genetic types of the Permian Prikam’e oils on the basis of their carbon isotope composition. Geol. Nefti Gaza 1972, 1, 33–39. [Google Scholar]
- Galimov, E.M.; Posyagin, V.I.; Prokhorov, V.S. Experimental study of carbon isotope fractionation in the CH4–C2H6–C3H8–C4H10 system at different temperatures. Geokhimiya 1972, 8, 977–987. [Google Scholar]
- Galimov, E.M.; Migdisov, A.A.; Ronov, A.B. Controlling factors of carbon isotope composition in the Precambrian and Phanerozoic. In Litologiya Osadochnaya Geologiya Dokembriya (Precambrian Lithology and Sedimentary Geology); Sidorenko, A.V., Ed.; Akademiya Nauk USSR: Moscow, Russia, 1973; pp. 279–281. [Google Scholar]
- Galimov, E.M.; Teplinskiy, G.I.; Tabassaranskiy, Z.A.; Gavrilov, Y.Y. On the conditions of formation of gas deposits in the eastern part of the Turan Plate as revealed by carbon isotopic composition of the gases. Geochem. Int. 1973, 10, 1259–1271. [Google Scholar]
- Galimov, E.M. Carbon Isotopes in Oil and Gas Geology; Nedra Press: Moscow, Russia, 1975; p. 385. [Google Scholar]
- Lowenstein, T.K.; Timofeeff, M.N.; Kovalevych, V.M.; Horita, J. The major-ion composition of Permian seawater. Geochim. Cosmochim. Acta 2005, 69, 1701–1719. [Google Scholar] [CrossRef]
- Zerkle, A.L.; Kamyshny, A.; Kump, L.R.; Farquhar, J.; Oduro, H.; Arthur, M.A. Sulfur cycling in a stratified euxinic lake with moderately high sulfate: Constraints from quadruple S isotopes. Geochim. Cosmochim. Acta 2010, 74, 4953–4970. [Google Scholar] [CrossRef]
- Johnston, D.T. Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle. Earth Sci. Rev. 2011, 106, 161–183. [Google Scholar] [CrossRef]
- Canfield, D.E. Biogeochemistry of sulfur isotopes. In Stable Isotope Geochemistry; Valley, J.W., Cole, D., Eds.; Mineralogical Society of America and Geochemical Society, Reviews of Mineralogy and Geochemistry: Washington, DC, USA, 2001; Volume 43, pp. 607–636. [Google Scholar]
- Hurtgen, M.T.; Arthur, M.A.; Suits, N.S.; Kaufman, A.J. The sulfur isotopic composition of Neoproterozoic seawater sulfate: Implications for asnowball Earth? Earth Planet. Sci. Lett. 2002, 203, 413–430. [Google Scholar] [CrossRef]
- Berner, R.A.; Raisswell, R. Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time: A new theory. Geochim. Cosmochim. Acta 1983, 47, 855–862. [Google Scholar] [CrossRef]
- Kleeberg, A. Interactions between bentic phosphorus release and sulfur cycling in Lake Scharmützelsee (Germany). Water Air Soil Pollut. 1997, 99, 391–399. [Google Scholar] [CrossRef]
- Sim, M.S.; Ono, S.; Donovan, K.; Templer, S.P.; Bosak, T. Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibriosp. Geochim. Cosmochim. Acta 2011, 75, 4244–4259. [Google Scholar] [CrossRef]
- Urey, H.C. The thermodynamic properties of isotopic substances. J. Chem. Soc. 1947, 1, 562–581. [Google Scholar] [CrossRef]
- Stahl, W.J. Carbon and nitrogen isotopes in hydrocarbon re-search and exploration. Chem. Geol. 1977, 20, 121–149. [Google Scholar] [CrossRef]
- Fuex, A. The use of stable carbon isotopes in hydrocarbon exploration. J. Geochem. Explor. 1977, 7, 155–188. [Google Scholar] [CrossRef]
- Schoell, M. Genetic characterization of natural gases. Am. Assoc. Pet. Geol. Bull. 1983, 67, 2225–2238. [Google Scholar]
- Schoell, M. Recent advances in petroleum isotope geochemistry. Org. Geochem. 1984, 6, 645–663. [Google Scholar] [CrossRef]
- Schoell, M. Stable isotopes in petroleum research. In Advances in Petroleum Geochemistry; Brooks, J., Welte, D., Eds.; Academic Press: London, UK, 1984; Volume 1, pp. 215–245. [Google Scholar]
- Elsner, M.; Zwank, L.; Hunkeler, D.; Schwarzenbach, R.P. A New Concept Linking Observable Stable Isotope Fractionation to Transformation Pathways of Organic Pollutants. Environ. Sci. Technol. 2005, 39, 6896–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood Lollar, B.; Slater, G.F.; Sleep, B.; Witt, M.; Kledka, G.M.; Harkness, M.; Spivack, J. Stable carbon isotope evidence for intrinsic bioremediation of tetrachloroethene and trichloroethene at area 6, Dover Air Force Base. Environ. Sci. Technol. 2001, 35, 261–269. [Google Scholar] [CrossRef]
- George, S.C.; Boreham, C.J.; Minifie, S.A.; Teerman, S.C. The effect of minor to moderate biodegradation on C5 to C9 hydrocarbons in crude oils. Org. Geochem. 2002, 33, 1293–1317. [Google Scholar] [CrossRef]
- Schüth, C.; Taubald, H.; Bolaño, N.; Maciejczyk, K. Carbon and hydrogen isotope effects during sorption of organic contaminants on carbonaceous materials. J. Contam. Hydrol. 2003, 64, 269–281. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; Georgi, A.; Voskamp, M.; Richnow, H.H. Carbon Isotope Fractionation of Organic Contaminants Due to Retardation on Humic Substances: Implications for Natural Attenuation Studies in Aquifers. Environ. Sci. Technol. 2005, 39, 6052–6062. [Google Scholar] [CrossRef]
- Kuder, T.; Philp, P.; Allen, J. Effects of Volatilization on Carbon and Hydrogen Isotope Ratios of MTBE. Environ. Sci. Technol. 2009, 43, 1763–1768. [Google Scholar] [CrossRef]
- Onojake, M.C.; Osuji, L.C.; Oforka, N.C. Bulk Geochemical Characteristics of Crude Oils From the Umutu/Bomu Fields, Niger Delta, Nigeria. Pet. Sci. Technol. 2014, 32, 618–624. [Google Scholar] [CrossRef]
- Murphy, M.T.K. Analytical methods. In Organic Geochemistry; Eglinton, G., Murphy, M.T.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1969; pp. 74–88. [Google Scholar]
- Hertelendi, E.; Gál, J.; Paál, A.; Fekete, S.; Giurgiu, M.; Gál, I.; Kertész, Z.; Nagy, S. Stable isotope mass spectrometer. In Proceedings of the Fourth Working Meeting Isotopes in Nature, Leipzig, Germany, 22–26 September 1986; pp. 323–334. [Google Scholar]
- Amrani, A.; Sessions, A.L.; Adkins, J.F. Compound-Specific δ34S Analysis of Volatile Organics by Coupled GC/Multicollector-ICPMS. Anal. Chem. 2009, 81, 9027–9034. [Google Scholar] [CrossRef] [PubMed]
- Amrani, A.; Deev, A.; Sessions, A.; Tang, Y.; Adkins, J.F.; Hill, R.J.; Moldowan, J.; Wei, Z. The sulfur-isotopic compositions of benzothiophenes and dibenzothiophenes as a proxy for thermochemical sulfate reduction. Geochim. Cosmochim. Acta 2012, 84, 152–164. [Google Scholar] [CrossRef]
- Said-Ahmad, W.; Amrani, A. A sensitive method for the sulfur isotope Analysis of dimethyl sulfide and dimethyl sulfoniopropionate in seawater. Rapid Commun. Mass Spectrom. 2013, 27, 2789–2796. [Google Scholar] [CrossRef]
- Giesemann, A.; Jäger, H.A.; Norman, A.L.; Krouse, H.R.; Brand, W.A. On-line sulphur isotope determination using an elemental analyzer coupled to a mass spectrometer. Anal. Chem. 1994, 66, 2816–2819. [Google Scholar] [CrossRef]
- Puchelt, H.; Sabels, B.R.; Hoering, T.C. Preparation of sulfur hexafluoride for isotope geochemical analysis. Geochim. Cosmochim. Acta 1971, 35, 625–628. [Google Scholar] [CrossRef]
- Rees, C. Sulphur isotope measurements using SO2 and SF6. Geochim. Cosmochim. Acta 1978, 42, 383–389. [Google Scholar] [CrossRef]
- Beaudoin, G.; Taylor, B.E. High precision and spatial resolution sulfur isotope analysis using MILES laser microprobe. Geochim. Cosmochim. Acta 1994, 58, 5055–5063. [Google Scholar] [CrossRef]
- Hu, G.X.; Rumble, D.; Wang, P.L. An ultraviolet laser microprobe for the in-situ analysis of multisulfur isotopes and its use in measuring Archean sulphur isotope mass-independent anomalies. Geochim. Cosmochim. Acta 2003, 67, 3101–3118. [Google Scholar] [CrossRef]
- Kelley, K.D.; Wilkinson, J.J.; Chapman, J.B.; Crowther, H.L.; Weiss, D. Zinc isotopes in sphalerite from base metal deposits in the red dog district, northern alaska. Econ. Geol. 2009, 104, 767–773. [Google Scholar] [CrossRef]
- Crowe, D.; Valley, J.W.; Baker, K.L. Micro-analysis of sulfur-isotope ratios and zonation by laser microprobe. Geochim. Cosmochim. Acta 1990, 54, 2075–2092. [Google Scholar] [CrossRef]
- Ono, S.; Wing, B.A.; Johnston, D.; Farquhar, J.; Rumble, D. Mass-dependent fractionation of quadruple sulphur isotope system as a new tracer of sulphur biogeochemical cycles. Geochim. Cosmochim. Acta 2006, 70, 2238–2252. [Google Scholar] [CrossRef]
- Chaussidon, M.; Albarede, F.; Sheppard, S.M.F. Sulphur isotope heterogeneity in the mantle from ion microprobe measurements of sulphide inclusions in diamonds. Nature 1987, 330, 242–244. [Google Scholar] [CrossRef]
- Chaussidon, M.; Albarede, F.; Sheppard, S.M.F. Sulphur isotope variations in the mantle from ion microprobe analysis of microsulphide inclusions. Earth Planet. Sci. Lett. 1989, 92, 144–156. [Google Scholar] [CrossRef]
- Eldridge, C.S.; Compston, W.; Williams, I.S.; Both, R.A.; Walshe, J.L.; Ohmoto, H. Sulfur isotope variability in sediment hosted massive sulfide deposits as determined using the ion microprobe SHRIMP. I. An example from the Rammelsberg ore body. Econ. Geol. 1988, 83, 443–449. [Google Scholar] [CrossRef]
- Eldridge, C.S.; Williams, I.S.; Walshe, J.L. Sulfur isotope variability in sediment hosted massive sulfide deposits as determined using the ion microprobe SHRIMP. II. A study of the H.Y.C. deposit at McArthur River, Northern Territory, Australia. Econ. Geol. 1993, 88, 1–26. [Google Scholar] [CrossRef]
- Kozdon, R.; Kita, R.N.; Huberty, J.M.; Fournelle, J.H.; Johnson, C.A.; Valley, J.W. In situ sulfur isotope analysis of sulfide minerals by SIMS: Precision and accuracy with application to thermometry of similar to 3.5 Ga Pilbara cherts. Chem. Geol. 2010, 275, 243–253. [Google Scholar] [CrossRef]
- Hauri, E.H.; Papineau, D.; Wang, J.; Hillion, F. High-precision analysis of multiple sulfur isotopes using NanoSIMS. Chem. Geol. 2016, 420, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Bendall, C.; Lahaye, Y.; Fiebig, J.; Weyer, S.; Brey, G.P. In situ sulfur isotope analysis by laser ablation MC-ICPMS. Appl. Geochem. 2006, 21, 782–787. [Google Scholar] [CrossRef]
- Craddock, P.R.; Rouxel, O.J.; Ball, L.A.; Bach, W. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chem. Geol. 2008, 253, 102–113. [Google Scholar] [CrossRef]
- Paris, G.; Sessions, A.; Subhas, A.V.; Adkins, J.F. MC-ICP-MS measurement of δ34 S and ∆33S in small amounts of dissolved sulphate. Chem. Geol. 2013, 345, 50–61. [Google Scholar] [CrossRef]
- Grotheer, H.; Greenwood, P.; McCulloch, M.; Böttcher, M.E.; Grice, K. δ34S character of organosulfur compounds in kerogen and bitumen fractions of sedimentary rocks. Org. Geochem. 2017, 110, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.E.; Fowler, M.G. Applications of petroleum geochemistry to exploration and reservoir management. Org. Geochem. 2002, 33, 5–36. [Google Scholar] [CrossRef]
- Craig, H. The geochemistry of the stable carbon isotopes. Geochim. Cosmochim. Acta 1953, 3, 53–92. [Google Scholar] [CrossRef]
- Silverman, S.R. Migration and segregation of oil and gas fluids in subsurface environments. Am. Assoc. Pet. Geol. Memoir. 1965, 4, 54–65. [Google Scholar]
- Andrusevich, V.E.; Engel, M.H.; Zumberge, J.E.; Brothers, L.A. Secular, episodic changes in stable carbon isotope composi-tion of crude oils. Chem. Geol. 1998, 152, 59–72. [Google Scholar] [CrossRef]
- Odden, W.; Barth, T.; Talbot, M. Compound-specific carbon isotope analysis of natural and artificially generated hydro-carbons in source rocks and petroleum fluids from offshore Mid-Norway. Org. Geochem. 2002, 33, 47–65. [Google Scholar] [CrossRef]
- Welte, D.; Hagemann, H.; Hollerbach, A.; Leythauser, D.; Stahl, W. Correlation between petroleum and source rock. In Proceedings of Ninth World Petroleum Congress; Applied Science Publishers II: London, UK, 1975; pp. 179–191. [Google Scholar]
- Philp, R.P.; Jarde, E. Application of stable and radioiso-topes. In Introduction to Environmental Forensics; Murphy, B., Morrison, R., Eds.; Elsevier: New York, NY, USA, 2007; pp. 455–512. [Google Scholar]
- Stahl, W. Source rock-crude oil correlation by isotopic type-curves. Geochim. Cosmochim. Acta 1978, 42, 1573–1577. [Google Scholar] [CrossRef]
- Chung, H.; Brand, S.W.; Grizzle, P.L. Carbon isotope geochemistry of Paleozoic oils from Big Horn Basin. Geochim. Cosmochim. Acta 1981, 45, 1803–1815. [Google Scholar] [CrossRef]
- Palmer, S.E. Effect of biodegradation and water-washing on crude oil composition. In Organic Geochemistry; Engel, M.H., Macko, S.E., Eds.; Plenum: New York, NY, USA, 1993; pp. 511–534. [Google Scholar]
- Stahl, W. Compositional changes and 13C/12C fractionations during the degradation of hydrocarbons by bacteria. Geochim. Cosmochim. Acta 1980, 44, 1903–1907. [Google Scholar] [CrossRef]
- Clayton, C. Carbon isotope fractionation during natural gas generation from kerogen. Mar. Pet. Geol. 1991, 8, 232–240. [Google Scholar] [CrossRef]
- Clayton, C. Effect of maturity on carbon isotope ratios of oils and condensates. Org. Geochem. 1991, 17, 887–899. [Google Scholar] [CrossRef]
- Hunt, J.M. Petroleum Geochemistry and Geology; W.H. Freeman: New York, NY, USA, 1996. [Google Scholar]
- Park, R.; Epstein, S. Carbon isotope fractionation during photosynthesis. Geochim. Cosmochim. Acta 1960, 21, 110–126. [Google Scholar] [CrossRef]
- O’Leary, M.H. Carbon isotope fractionation in plants. Phytochemistry 1981, 20, 553–567. [Google Scholar] [CrossRef]
- Stuermer, D.H.; Peters, K.; Kaplan, I. Source indicators of humic substances and proto-kerogen. Stable isotope ratios, elemental compositions and electron spin resonance spectra. Geochim. Cosmochim. Acta 1978, 42, 989–997. [Google Scholar] [CrossRef]
- Aizenshtat, Z.; Baedecker, M.; Kaplan, I. Distribution and diagenesis of organic compounds in JOIDES sediment from Gulf of Mexico and western Atlantic. Geochim. Cosmochim. Acta 1973, 37, 1881–1898. [Google Scholar] [CrossRef]
- Sackett, W.M.; Eadie, B.J.; Exner, M.E. Stable isotope com-position of organic carbon in recent Antarctic sediments. In Advances in Organic Geochemistry; Tissot, B., Bienner, F., Eds.; Technip: Paris, France, 1974; pp. 661–671. [Google Scholar]
- Gormley, J.R.; Sackett, W.M. Carbon isotope evidence for the maturation of marine lipids. In Advances in Organic Geochemistry; Campos, R., Goni, J., Eds.; Enadisma: Madrid, Spain, 1977; pp. 321–339. [Google Scholar]
- Brown, F.; Baedecker, M.; Nissenbaum, A.; Kaplan, I. Early diagenesis in a reducing fjord, Saanich Inlet, British Columbia—III. Changes in organic constituents of sediment. Geochim. Cosmochim. Acta 1972, 36, 1185–1203. [Google Scholar] [CrossRef]
- Peters, K.E.; Rohrback, B.G.; Kaplan, I.R. Carbon and hydro-gen stable isotope variations in kerogen during laboratory-simulated thermal maturation. Am. Assoc. Pet. Geol. Bull. 1981, 65, 501–508. [Google Scholar]
- Hatcher, P.G.; Spiker, E.C.; Szeverenyi, N.M.; Maciel, G.E. Selective preservation and origin of petroleum-forming aquatic kerogen. Nature 1983, 305, 498–501. [Google Scholar] [CrossRef]
- Hunt, J. The significance of carbon isotope variations in marine sediments. In Advances in Organic Geochemistry; Hobson, G.D., Speers, G.C., Eds.; Pergamon: Oxford, UK, 1970. [Google Scholar]
- Mello, M.R.; Telnaes, N.; Gaglianone, P.C.; Chicarelli, M.I.; Brassell, S.C.; Maxwell, J.R. Organic geochemical characterization of depositional palaeoenvironments of source rocks and oils in Brazilian marginal basins. Org. Geochem. 1988, 13, 31–45. [Google Scholar] [CrossRef]
- Tissot, B.; Welte, D. Petroleum Formation and Occurrence, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1984. [Google Scholar]
- Sun, Y.; Chen, Z.; Xu, S.; Cai, S. Stable carbon and hydrogen isotopic fractionation of individual n-alkanes accompanying biodegradation: Evidence from a group of progressively bio-degraded oils. Org. Geochem. 2005, 36, 225–238. [Google Scholar] [CrossRef]
- Peters, K.E.; Walters, C.C.; Moldowan, J.M. The Biomarker Guide; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Boreham, C.J.; Dowling, L.M.; Murray, A.P. Biodegradation and maturity influences on n-alkane isotopic profiles in terrigenous sequences. In Proceedings of the 17th International Meeting on Organic Geochemistry, San Sebastian, Spain, 4–8 September 1995; pp. 539–541. [Google Scholar]
- Mansuy, L.; Philp, R.P.; Allen, J. Source identification of oil spills based on the isotopic compositin of individual compo-nents in weathered oil samples. Environ. Sci. Technol. 1997, 31, 3417–3425. [Google Scholar] [CrossRef]
- Huang, Y.; Eglinton, G.; Ineson, P.; Latter, P.; Bol, R.; Harkness, D. Absence of carbon isotope fractionation of individual n-alkanes in a 23-year field decomposition experiment with Calluna vulgaris. Org. Geochem. 1997, 26, 497–501. [Google Scholar] [CrossRef]
- Wilkes, H.; Boreham, C.; Harms, G.; Zengler, K.; Rabus, R. Anaerobic degradation and carbon isotopic fractionation of alkylbenzenes in crude oil by sulphate-reducing bacteria. Org. Geochem. 2000, 31, 101–115. [Google Scholar] [CrossRef]
- Hunkeler, D.; Andersen, N.; Aravena, R.; Bernasconi, S.M.; Butler, B.J. Hydrogen and Carbon Isotope Fractionation during Aerobic Biodegradation of Benzene. Environ. Sci. Technol. 2001, 35, 3462–3467. [Google Scholar] [CrossRef] [Green Version]
- Masterson, W.D.; Dzou, L.I.P.; Holba, A.G.; Fincannon, A.L.; Ellis, L. Evidence for biodegradation and evaporative frac-tionation in West Sak, Kuparuk and Prudhoe Bay field areas, North Slope, Alaska. Org. Geochem. 2001, 32, 411–441. [Google Scholar] [CrossRef]
- Mazeas, L.; Budzinski, H.; Raymond, N. Absence of stable carbon isotopic fractionation of saturated and polycyclic aromatic hydrocarbons during aerobic bacterial biodegrada-tion. Org. Geochem. 2002, 33, 1259–1272. [Google Scholar] [CrossRef]
- Pond, K.L.; Huang, Y.; Wang, Y.; Kulpa, C.F. Hydrogen isotopic composition of individual n-alkanes as an intrinsic tracer for bioremediation and source identification of petro-leum contamination. Environ. Sci. Technol. 2002, 36, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.A.; Ulrich, A.C.; Lacrampe-Couloume, G.; Sleep, B.; Edwards, E.A.; Lollar, B.S. Carbon and Hydrogen Isotopic Fractionation during Anaerobic Biodegradation of Benzene. Appl. Environ. Microbiol. 2003, 69, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallasser, R.J. Recognizing biodegradation in gas/oil accu-mulations through the δ13C compositions of gas components. Org. Geochem. 2000, 31, 1363–1373. [Google Scholar] [CrossRef]
- Hoeffs, J.; Frey, M. The isotopic composition of carbona-ceous matter in metamorphic profile from the Swiss Alps. Geochim. Cosmochim. Acta 1976, 40, 945–951. [Google Scholar] [CrossRef]
- Lewan, M. Effects of thermal maturation on stable organic carbon isotopes as determined by hydrous pyrolysis of Woodford Shale. Geochim. Cosmochim. Acta 1983, 47, 1471–1479. [Google Scholar] [CrossRef]
- Lewan, M.; Winters, J.; McDonald, J. Generation of oil-like pyrolysates from organic-rich shales. Science 1979, 203, 896–899. [Google Scholar] [CrossRef]
- Lewan, M.; Williams, J. Evaluation of petroleum generation from resinites by hydrous pyrolysis. Am. Assoc. Pet. Geol. Bull. 1987, 71, 207–214. [Google Scholar]
- Moldowan, J.M.; Dahl, J.E.; Huizinga, B.J.; Jacobson, S.R.; Taylor, D.W. The relationship of angiosperms and oleanane in petroleum through geologic time. Am. Assoc. Pet. Geol. Bull. 1993, 77, 334–335. [Google Scholar]
- Chung, H.M.; Rooney, M.A.; Toon, M.B.; Claypool, G.E. Carbon isotope composition of marine crude oils. Bull. Am. Assoc. Pet. Geol. 1992, 76, 1000–1007. [Google Scholar]
- Knoll, A.H. End of the Proterozoic Eon. Sci. Am. 1991, 265, 64–73. [Google Scholar] [CrossRef]
- Matthews, D.E.; Hayes, J.M. Isotope-ratio-monitoring gas chromatography-mass spectrometry. Anal. Chem. 1978, 50, 1465–1473. [Google Scholar] [CrossRef]
- Hayes, J.M.; Takigiku, R.; Ocampo, R.; Callot, H.J.; Albrecht, P. Isotopic composition and probable origins of organic molecules in the Eocene Messel Shale. Nature 1987, 329, 48–51. [Google Scholar] [CrossRef]
- Hayes, J.M.; Freeman, K.H.; Popp, B.N.; Hoham, C.H. Compound-specific isotopic analysis: A novel tool for reconstruction of ancient biogeochemical processes. Org. Geochem. 1990, 16, 1115–1128. [Google Scholar] [CrossRef]
- Freeman, K.; Hayes, J.M.; Trendel, J.-M.; Albrecht, P. Evidence from carbon isotope measurements for diverse origins of sedimentary hydrocarbons. Nature 1990, 343, 254–256. [Google Scholar] [CrossRef]
- Philp, R.P. Reservoir Geochemistry; The University of Oklahoma: Norman, OK, USA, 1996. [Google Scholar]
- Rincon, J.; Jaramillo, J.; Philp, R.; Allen, J. Biomar-kers and compound-specific stable carbon isotope of n-alkanes in crude oils from Eastern Llanos Basin, Colombia. J. S. Am. Earth Sci. 2010, 29, 198–213. [Google Scholar]
- Liu, J.; Geng, A.; Xiong, Y. The application of stable carbon and hydrogen isotopic compositions of individual n-alkanes to Paleozoic oil/source rock correlation enigmas in the Huanghua depression, China. J. Pet. Sci. Eng. 2006, 54, 70–78. [Google Scholar] [CrossRef]
- Bjorøy, M.; Hall, K.; Gillyon, P.; Jumeau, J. Carbon isotope variations in n-alkanes and isoprenoids of whole oils. Chem. Geol. 1991, 93, 13–20. [Google Scholar] [CrossRef]
- Karlsen, D.A.; Nyland, B.; Flood, B.; Ohm, S.E.; Brekke, T.; Olsen, S.; Backer-Owe, K. Petroleum geochemistry of the Haltenbanken, Norwegian continental shelf. In The Geochemistry of Reservoirs Geological Society; Cubbit, J.M., England, W.A., Eds.; Special Publication: London, UK, 1995; pp. 203–256. [Google Scholar]
- Stoddart, D.P.; Hall, P.B.; Larter, S.R.; Brasher, J.; Li, M.; Bjorøy, M. The reservoir geochemistry of the Eldfisk Field, Norwegian North Sea. In The Geochemistry of Reservoirs Geological Society; Cubitt, J.M., England, W.A., Eds.; Special Publication: London, UK, 1995; pp. 257–279. [Google Scholar]
- Schoell, M.; Hwang, R.; Carlson, R.; Welton, J. Carbon isotopic composition of individual biomarkers in gilsonites (Utah). Org. Geochem. 1994, 21, 673–683. [Google Scholar] [CrossRef]
- Rooney, M.; Vuletich, A.; Griffith, C. Compound-specific isotope analysis as a tool for characterizing mixed oils: An example from the West of Shetlands area. Org. Geochem. 1998, 29, 241–254. [Google Scholar] [CrossRef]
- Inaba, T.; Suzuki, N. Gel permeation chromatography for fractionation and isotope ratio analysis of steranes and tri-terpanes in oils. Org. Geochem. 2003, 34, 635–641. [Google Scholar] [CrossRef]
- Riediger, C.; Fowler, M.; Snowdon, L. Organic geochemis-try of the Lower Cretaceous Ostracode Zone, a brackish/non-marine source for some lower Manville oils in southeastern Alberta. Can. Soc. Pet. Geol. Mem. 1997, 18, 93–102. [Google Scholar]
- Schaeffer, P.; Poinsot, J.; Hauke, V.; Adam, P.; Wehrung, P.; Trendel, J.M.; Albrecht, P.; Dessort, D.; Connan, J. Novel optically active hydrocarbons in sediments: Evidence for an extensive biological cyclization of higher regular polyphenols. Angew. Chem. Int. 1994, 33, 1166–1169. [Google Scholar] [CrossRef]
- Li, M.; Riediger, C.; Fowler, M.; Snowdon, L. Unusual polycyclic aromatic hydrocarbons in Lower Cretaceous Ostracode Zone sediments and related oils of the Western Canada sedimentary basin. Org. Geochem. 1997, 27, 439–448. [Google Scholar] [CrossRef]
- Cuishan, Z.; Hong, Z.; Peirong, W.; Zebo, Z.; Anding, C. The distribution and carbon isotopic composition of unusual polycyclic alkanes in the Cretaceous Lengshuiwu Formation, China. Org. Geochem. 2003, 34, 1027–1035. [Google Scholar] [CrossRef]
- Li, M.; Huang, Y.S.; Obermajer, M.; Jiang, C.Q.; Snowdon, L.R.; Fowler, M.G. Hydrogen isotopic compositions of individual alkanes as a new approach to petroleum correlation:case studies from the Western Canada Sedimentary Basin. Org. Geochem. 2001, 32, 1387–1399. [Google Scholar] [CrossRef]
- Xiong, Y.; Geng, A. Carbon isotopic composition of indi-vidual n-alkanes in asphaltene pyrolysates of biodegraded crude oils from the Liaohe basin, China. Org. Geochem. 2000, 31, 1441–1449. [Google Scholar] [CrossRef]
- Liao, Y.; Geng, A.; Xiong, Y.; Liu, D.; Lu, J.; Liu, J.; Zhang, H.; Geng, X. The influence of hydrocarbon expulsion on carbon isotopic compositions of individual n-alkanes in pyrolysates of selected terrestrial kerogens. Org. Geochem. 2004, 35, 1479–1488. [Google Scholar] [CrossRef]
- Xiong, Y.; Geng, A.; Pan, C.; Liu, D.; Peng, P. Characterization of the hydrogen isotopic composition of individual n-alkanes in terrestrial source rocks. Appl. Geochem. 2005, 20, 455–464. [Google Scholar] [CrossRef]
- Li, M.; Larter, S.; Mei, B.; Bjorøy, M. Compound specific isotopic compositions for end members of crude oils and related source rocks from the Liaohe Basin: Paleoenvironmental implications. In Organic Geochemistry: Developments and Application to Energy, Climate and Human History, Proceedings of the 17th International Meeting on Organic Geochemistry, San Sebastián, Spain, 4–8 September 1995; Grimalt, J.O., Dorronsoro, C., Eds.; A.I.G.O.A.: San Sebastián, Spain, 1995; pp. 38–40. [Google Scholar]
- Xiong, Y.; Geng, A.; Wang, C.; Sheng, G.; Fu, J. The origin of crude oils from the Shuguang-Huanxiling Buried Hills in the Liaohe Basin, China: Evidence from chemical and isotopic compositions. Appl. Geochem. 2003, 18, 445–456. [Google Scholar] [CrossRef]
- Bernard, B.; Brooks, J.; Sackett, W. Natural gas seepage in the Gulf of Mexico. Earth Planet. Sci. Lett. 1976, 31, 48–54. [Google Scholar] [CrossRef]
- Gurgey, K.; Philp, P.; Clayton, C.; Emiroglu, H.; Siyako, M. Geochemical and isotopic approach to maturity/source/mixing estimations for natural gas and associated condensates in the Thrace Basin, NW Turkey. Appl. Geochem. 2005, 20, 2017–2037. [Google Scholar] [CrossRef]
- Tuo, J.; Zhang, M.; Wang, X.; Zhang, C. Hydrogen isotope ratios of aliphatic and diterpenoid hydrocarbons in coals and carbonaceous mudstones from the Liaohe Basin, China. Org. Geochem. 2006, 37, 165–176. [Google Scholar] [CrossRef]
- Alexander, R.; Kagi, R.I.; Larcher, A.V. Clay catalysis of alkyl hydrogen exchange reactions—reaction mechanisms. Org. Geochem. 1984, 6, 755–760. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Lewan, M.D.; Wintsch, R.P. D/H isotope ratios of kerogen, bitumen, oil, and water in hydrous pyrolysis of source rocks containing kerogen types I.; II, IIS, and III. Geochim. Cosmochim. Acta 1999, 63, 3751–3766. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Boudou, J.-P.; Lewan, M.D.; Wintsch, R.P. Experimental controls on D/H and 13C/12C ratios of kerogen, bitumen and oil during hydrous pyrolysis. Org. Geochem. 2001, 32, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Leif, R.N.; Simoneit, B.R. The role of alkenes produced during hydrous pyrolysis of a shale. Org. Geochem. 2000, 31, 1189–1208. [Google Scholar] [CrossRef]
- Sessions, A.L. Hydrogen Isotope Ratios of Individual Organic Compounds. Ph.D. Thesis, Indiana University, Bloomington, Indiana, 2001; p. 149. [Google Scholar]
- Sessions, A.L.; Sylva, S.P.; Summons, R.E.; Hayes, J.M. Isotopic exchange of carbon-bound hydrogen over geologic timescales. Geochim. Cosmochim. Acta 2004, 68, 1545–1559. [Google Scholar] [CrossRef]
- Pedentchouk, N.; Freeman, K.H.; Harris, N.B. Different response of δD values of n-alkanes, isoprenoids, and kerogen during thermal maturation. Geochim. Cosmochim. Acta 2006, 70, 2063–2072. [Google Scholar] [CrossRef]
- Stalker, L.; Larter, S.R.; Farrimond, P. Biomarker binding into keogens: Evidence from hydrous pyrolysis using heavy water (D2O). Org. Geochem. 1998, 28, 239–253. [Google Scholar] [CrossRef]
- Burgoyne, T.W.; Hayes, J.M. Quantitative Production of H2 by Pyrolysis of Gas Chromatographic Effluents. Anal. Chem. 1998, 70, 5136–5141. [Google Scholar] [CrossRef]
- Hilkert, A.W.; Douthitt, C.B.; Schlüter, H.J.; Brand, W.A. Isotope ratio monitoring gas chromatography/Mass spectrometry of D/H by high temperature conversion isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 1226–1230. [Google Scholar] [CrossRef]
- Sessions, A.; Burgoyne, T.W.; Schimmelmann, A.; Hayes, J.M. Fractionation of hydrogen isotopes in lipid biosynthesis. Org. Geochem. 1999, 30, 1193–1200. [Google Scholar] [CrossRef]
- Anderson, K.B.; Muntean, J.V. The nature and fate of natural resins in the geosphere. Part, X. Structural characteristics of the macromolecular constituents of modern dammar resin and Class II ambers. Geochem. Trans. 2000, 1, 1–9. [Google Scholar] [CrossRef]
- Maslen, E.; Grice, K.; Le Métayer, P.; Dawson, D.; Edwards, D. Stable carbon isotopic compositions of individual aromatic hydrocarbons as source and age indicators in oils from western Australian basins. Org. Geochem. 2011, 42, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Andersen, N.; Paul, H.A.; Bernasconi, S.; McKenzie, J.A.; Behrens, A.; Schaeffer, P.; Albrecht, P. Large and rapid climate variability during the Messinian salinity crisis: Evidence from deuterium concentrations of individual biomarkers. Geology 2001, 29, 799–802. [Google Scholar] [CrossRef]
- Yang, H.; Huang, Y. Preservation of lipid hydrogen isotope ratios in Miocene lacustrine sediments and plant fossils at Clarkia, northern Idaho, USA. Org. Geochem. 2003, 34, 413–423. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R. Stable hydrogen isotopic composition of hydrocarbons in crude oils and source rocks from the Perth Basin (Western Australia). In Proceedings of the 21st International Meeting on Organic Geochemistry, Krakow, Poland, 8–12 September 2003. [Google Scholar]
- Dansgaard, W. Stable isotopes in precipitation. Tellus 1964, 16, 436–468. [Google Scholar] [CrossRef]
- Kehew, A.E. Applied Chemical Hydrogeology; Prentice Hall: Upper Saddle River, NJ, USA, 2001. [Google Scholar]
- Schoell, M. The hydrogen and carbon isotopic composition of methane from natural gases of various origins. Geochim. Cosmochim. Acta 1980, 44, 649–661. [Google Scholar] [CrossRef]
- Schoell, M. Multiple origins of methane in the earth. Chem. Geol. 1988, 71, 1–10. [Google Scholar] [CrossRef]
- Dai, J.; Pei, X.; Qi, H. China Natural Gas Geology; Petroleum Industry Press: Beijing, China, 1992; Volume 1, pp. 35–86. [Google Scholar]
- Dai, J.X.; Xia, X.Y.; Li, Z.S. Inter-laboratory calibration of natural gas round robins for δ2H and δ13C using offline and online techniques. Chem. Geol. 2012, 310–311, 49–55. [Google Scholar] [CrossRef]
- Dai, J.X.; Ni, Y.Y.; Gong, D.Y. Geochemical characteristics of gases of from the largest tight sand field (Sulige) and shale gas field (Fuling) in China. Mar. Pet. Geol. 2017, 79, 426–438. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Faber, E.; Schoell, M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation-isotope evidence. Geochim. Cosmochim. Acta 1986, 50, 693–709. [Google Scholar] [CrossRef]
- Whiticar, M.J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 1999, 161, 291–314. [Google Scholar] [CrossRef]
- Shen, P.; Xu, Y.C. Isotopic compositional characteristics of terrigenous natural gases in China. Chin. J. Geochem. 1993, 12, 14–24. [Google Scholar] [CrossRef]
- Xu, Y. Origin Theory and Application of Natural Gases; Science Press: Beijing, China, 1994. [Google Scholar]
- Galimov, E.M. Isotope organic geochemistry. Org. Geochem. 2006, 37, 1200–1262. [Google Scholar] [CrossRef]
- Kinnaman, F.S.; Valentine, D.L.; Tyler, S.C. Carbon and hydrogen isotope fractionation associated with aerobic microbial oxidation of methane, ethane, propane and butane. Geochim. Cosmochim. Acta 2007, 71, 271–283. [Google Scholar] [CrossRef]
- Ni, Y.Y.; Ma, Q.S.; Geoffrey, S.E. Fundamental studies on kinetic isotope effect (KIE) of hydrogen isotope fractionation in natural gas systems. Geochim. Cosmochim. Acta 2011, 75, 2696–2707. [Google Scholar] [CrossRef]
- Ni, Y.Y.; Dai, J.X.; Zhu, G.Y. Stable hydrogen and carbon isotopic ratios of coal-derived an oil-derived gases: A case study in the Tarim Basin, NW China. Int. J. Coal. Geol. 2013, 116–117, 302–313. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Worden, R.H.; Jin, Z.J. Thermochemical sulphate reduction(TSR) versus maturation and their effects on hydrogen stable isotopes of very dry alkane gases. Geochim. Cosmochim. Acta 2014, 137, 208–220. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Jin, Z.J.; Meng, Q.Q. Genetic types of natural gas and filling patterns in Daniudi gas field, Ordos Basin. China J. Asian Earth Sci. 2015, 107, 1–11. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Li, Z. The hydrogen isotopic characteristics of the Upper Paleozoic natural gas in Ordos Basin. Org. Geochem. 2014, 74, 66–75. [Google Scholar] [CrossRef]
- Wang, Y.P.; Dai, J.X.; Zhao, C.Y. Genetic origin of Mesozoic natural gases in the Ordos Basin (China): Comparison of carbon and hydrogen isotopes and pyrolytic results. Org. Geochem. 2010, 41, 1045–1048. [Google Scholar] [CrossRef]
- Dai, J.; Ni, Y.; Hu, G. Stable carbon and hydrogen isotopes of gases from the large tight gas fields in China. Sci. China Earth Sci. 2014, 57, 88–103. [Google Scholar] [CrossRef]
- Hu, G.Y.; Li, J.; Shan, X.Q. The origin of natural gas and the hydrocarbon charging history of the Yulin Gas field in the Ordos Basin, China. Int. J. Coal Geol. 2010, 81, 381–391. [Google Scholar]
- Hu, G.Y.; Yu, C.; Ni, Y.Y. Comparative study of stable carbon and hydrogen isotopes of alkane gases sourced from the Longtan and Xujiahe coal-bearing measures in the Sichuan Basin, China. Int. J. Coal Geol. 2014, 116–117, 293–301. [Google Scholar] [CrossRef]
- Wu, X.; Wang, P.; Liu, Q. The source of natural gas reservoired in the 5th member of the Upper Trias-sic Xujiahe Formation in Xinchang Gasfield, the Western Sichuan Depression and its implication. Nat. Gas Geosci. 2016, 27, 1409–1418. [Google Scholar]
- Huang, S.P.; Fang, X.; Liu, D. Natural gas genesis and sources in the Zizhou gas field, Ordos Basin, China. Int. J. Coal Geol. 2015, 152, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, J.; Li, Z. Characteristics and genetic types of the lower Paleozoic natural gas, Ordos Basin. Mar. Pet. Geol. 2018, 89, 106–119. [Google Scholar] [CrossRef]
- Wang, X.F.; Liu, W.H.; Shi, B.G. Hydrogen isotope characteristics of thermogenic methane in Chinese sedimentary basins. Org. Geochem. 2015, 83–84, 178–189. [Google Scholar] [CrossRef]
- Feng, Z.Q.; Liu, D.; Huang, S.P. Geochemical characteristics and genesis of natural gas in the Yan’an gas field, Ordos Basin, China. Org. Geochem. 2016, 102, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Q.; Tao, X.W.; Hu, G.Y. Geochemical characteristics and source of natural gases from southeast depression of the Tarim Basin, NW China. Org. Geochem. 2014, 74, 106–115. [Google Scholar] [CrossRef]
- Peng, W.; Hu, G.; Huang, S. Natural gas geochemical characteristics and genetic analysis: A case study of the Dongsheng gas field in the Ordos Basin of China. J. China Univ. Min. Technol. 2017, 46, 74–84. [Google Scholar]
- Huang, S.; Duan, S.; Wang, Z.; Jiang, Q.; Jiang, H.; Su, W.; Feng, Q.; Huang, T.; Yuan, M.; Ren, M.; et al. Affecting factors and application of the stable hydrogen isotopes of alkane gases. Pet. Explor. Dev. 2019, 46, 518–530. [Google Scholar] [CrossRef]
- Thode, H.G. Sulfur isotope ratios in petroleum research and exploration: 202 Williston basin. Am. Associ. Pet. Geol. Bull. 1981, 65, 1527–1535. [Google Scholar]
- Orr, W.L. Kerogen/asphaltene/sulphur relationships in sulphur-rich Montereyoils. In Advances in Organic Geochemistry; By, D., Leythaeuser, J., Eds.; Rullkotter Rullkotter Pergamon Press: Oxford, UK, 1986; pp. 499–516. [Google Scholar]
- Cai, C.F.; Zhang, C.M.; Cai, L.L.; Wu, G.H.; Jiang, L.; Xu, Z.M.; Li, K.K.; Ma, A.L.; Chen, L.X. Origins of Palaeozoic oils in the Tarim Basin: Evidence from sulfur isotopes and biomarkers. Chem. Geol. 2009, 268, 197–210. [Google Scholar] [CrossRef]
- Yan, D.T.; Chen, D.Z.; Wang, Q.C.; Wang, J.G. Predominance of stratified anoxic Yangtze Sea interrupted by short-term oxygenation during the Ordo-Silurian transition. Chem. Geol. 2012, 291, 69–78. [Google Scholar] [CrossRef]
- Wei, H.Y.; Wei, X.M.; Qiu, Z.; Song, H.Y.; Shi, G. Redox conditions across the G-L boundary in South China: Evidence from pyrite morphology and sulfur isotopic compositions. Chem. Geol. 2016, 440, 1–14. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Jin, Z.; Meng, Q.; Li, S. Influence of volcanic activities on redox chemistry changes linked to the enhancement of the ancient Sinian source rocks in the Yangtze craton. Precambrian Res. 2019, 327, 1–13. [Google Scholar] [CrossRef]
- Chen, G.; Chang, X.; Gang, W.; Wang, N.; Zhang, P.; Cao, Q.; Xu, J. Anomalous positive pyrite sulfur isotope in lacustrine black shale of the Yanchang Formation, Ordos Basin: Triggered by paleoredox chemistry changes. Mar. Pet. Geol. 2020, 104587. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of GC-IRMS system (modified from [21]).
Figure 1.
Schematic diagram of GC-IRMS system (modified from [21]).

Figure 2.
A plot of δ13C saturates versus δ13C aromatics (Reproduced from [91] with permission of Elsevier).
Figure 2.
A plot of δ13C saturates versus δ13C aromatics (Reproduced from [91] with permission of Elsevier).

Figure 3.
The isotopic relationship between the carbon isotope values of the various fractions and the source rock extracts. SAT: saturate hydrocarbons; OIL: whole oil; AROM: aromatic hydrocarbons; NSO’s: nitrogen, sulfur, oxygen compounds; ASPH: asphaltenes (Reproduced from [50] with permission from Elsevier).
Figure 3.
The isotopic relationship between the carbon isotope values of the various fractions and the source rock extracts. SAT: saturate hydrocarbons; OIL: whole oil; AROM: aromatic hydrocarbons; NSO’s: nitrogen, sulfur, oxygen compounds; ASPH: asphaltenes (Reproduced from [50] with permission from Elsevier).

Figure 4.
Saturate and aromatic carbon isotopic data of the crude oils from Brazil. (Reproduced from [109] with permission from Elsevier).
Figure 4.
Saturate and aromatic carbon isotopic data of the crude oils from Brazil. (Reproduced from [109] with permission from Elsevier).

Figure 5.
Changes in the isotopic compositions of crude oils and kerogen with maturation. (Reproduced from [124] with permission from Elsevier).
Figure 5.
Changes in the isotopic compositions of crude oils and kerogen with maturation. (Reproduced from [124] with permission from Elsevier).

Figure 6.
Stable carbon isotope composition of crude oils versus geologic age (Reproduced. from [88] with permission from Elsevier).
Figure 6.
Stable carbon isotope composition of crude oils versus geologic age (Reproduced. from [88] with permission from Elsevier).

Figure 7.
Carbon isotopic compositions of individual n-alkanes in source rocks of the.Western depression of the Liaohe Basin (Reproduced from [151] with permission from Elsevier).
Figure 7.
Carbon isotopic compositions of individual n-alkanes in source rocks of the.Western depression of the Liaohe Basin (Reproduced from [151] with permission from Elsevier).

Figure 8.
The relationship between carbon isotope values in methane and the C1/(C2+C3). compositional ratios of natural gas samples. (Reproduced from [154] with permission from Elsevier).
Figure 8.
The relationship between carbon isotope values in methane and the C1/(C2+C3). compositional ratios of natural gas samples. (Reproduced from [154] with permission from Elsevier).

Figure 9.
Diagram establishing the relationships between families of gases within a specific. Basin (after [21]).
Figure 9.
Diagram establishing the relationships between families of gases within a specific. Basin (after [21]).

Figure 10.
Plot showing the relationships between gases and condensates produced from the same formation (Reproduced from [155] with permission from Elsevier).
Figure 10.
Plot showing the relationships between gases and condensates produced from the same formation (Reproduced from [155] with permission from Elsevier).


{kind=link}
{kind=link}
{kind=link}
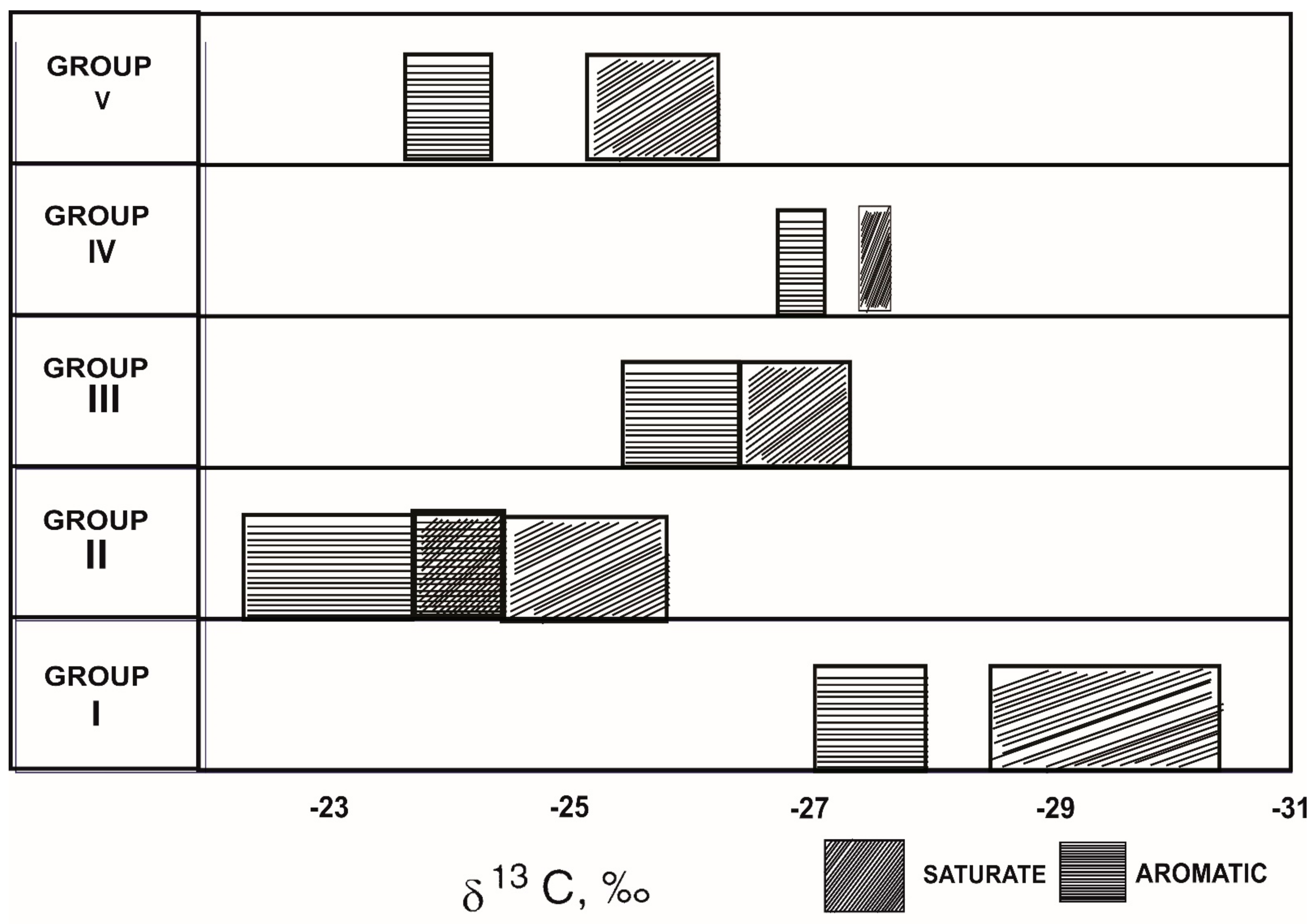{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
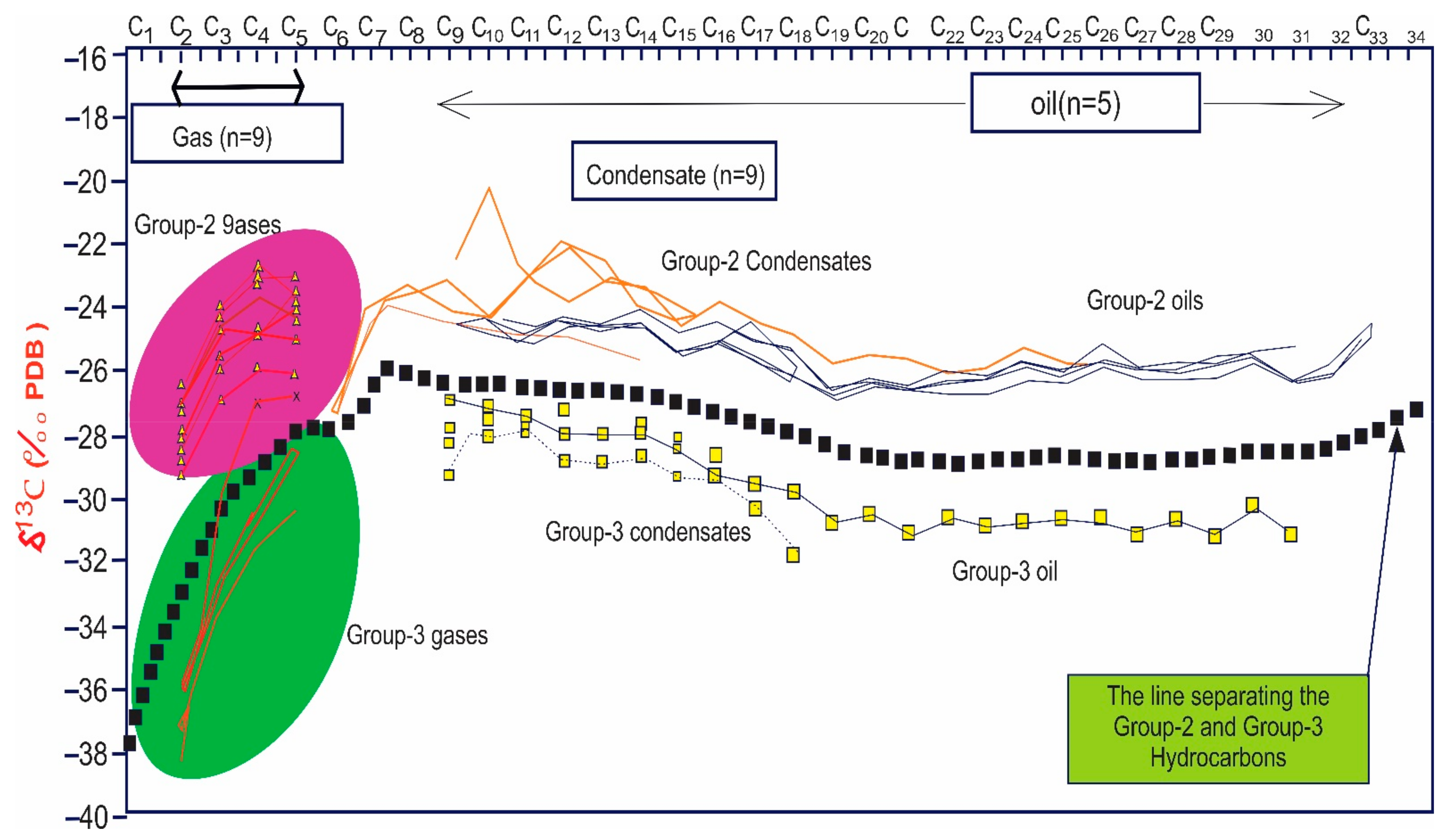{kind=link}
Table 1.
Isotopic ratios of stable isotopes [21].
Table 1.
Isotopic ratios of stable isotopes [21].
| Element | Notation | Ratio | Standard | Absolute Ratio |
|---|---|---|---|---|
| Hydrogen | δD | D/H(2H/1H) | SMOW | 1.557 × 10−4 |
| >Carbon | δ13C | 13C/12C | PDB | 1.122 × 10−2 |
| Nitrogen | δ15N | 15N/14N | Atmosphere | 3.613 × 10−3 |
| Oxygen | δ18O | 18O/16O | SMOW, PDB | 2.0052 × 10−3 |
| Chlorine | δ37Cl | 37Cl/35Cl | seawater | −0.31978 |
| Sulfur | δ34S | 34S/32S | CDT | 4.43 × 10−2 |
SMOW = standard mean ocean water; PDB = Pee Dee belemnite; CDT = Cañon Diablo troilite.
Table 2.
Bulk and elemental properties of Brazilian offshore oils (Reproduced from [109] with the permission of Elsevier).
Table 2.
Bulk and elemental properties of Brazilian offshore oils (Reproduced from [109] with the permission of Elsevier).
| Property | Group I | Group II | Group III | Group IV | Group V |
|---|---|---|---|---|---|
| δ13C‰ | < −28.0 | −23.0 to −25.6 | −25.4 to −26.6 | −26.8 to −27.6 | −24.4, −25.1 |
| % Sulphur | Low | Medium | High | High | Medium |
| V/Ni | Low | High/medium | Medium | High | High |
| %Saturates | High | High | Low/medium | Low/medium | High |
| n-Alkanes’ dominance | C23-C25 | C17-C21 | C17-C19 | C18-C20 | C20-C22 |
| Odd/even | High | High | Low | Low | Low |
| Pristane/Phytane | High | High | Low | Low | Low |
| Inferred depositional environment | Lacustrine/freshwater | Lacustrine/saline water | Marine evaporitic | Marine carbonate | Marine deltaic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ogbesejana, A.B.; Liu, B.; Ostadhassan, M. Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review. Molecules 2022, 27, 34. https://doi.org/10.3390/molecules27010034
AMA Style
Ogbesejana AB, Liu B, Ostadhassan M. Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review. Molecules. 2022; 27(1):34. https://doi.org/10.3390/molecules27010034
Chicago/Turabian StyleOgbesejana, Abiodun Busuyi, Bo Liu, and Mehdi Ostadhassan. 2022. "Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review" Molecules 27, no. 1: 34. https://doi.org/10.3390/molecules27010034