
ERK1/2 Activity Is Critical for the Outcome of Ischemic Stroke

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
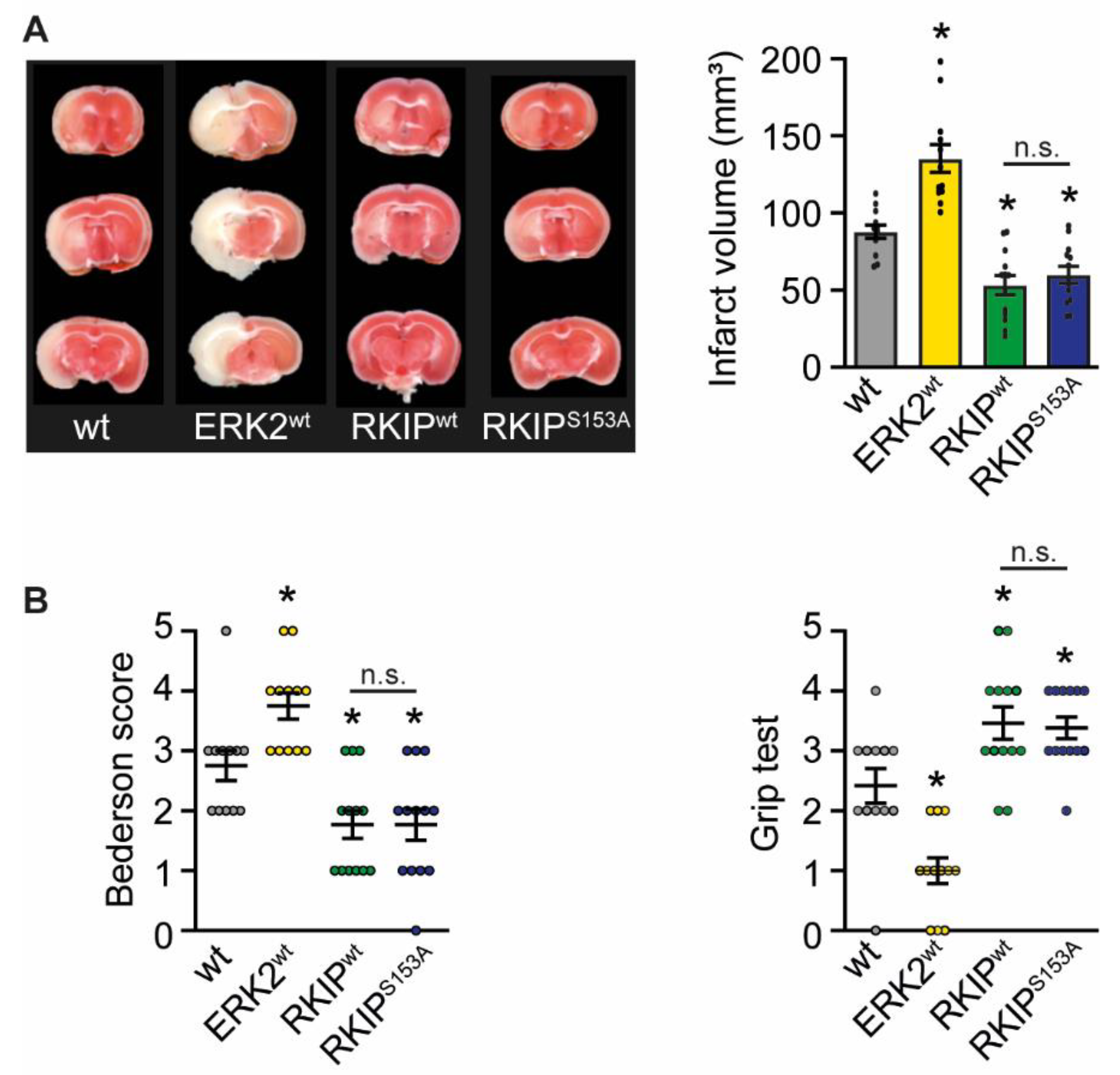
2.1. ERK2wt Overexpression Leads to Massive Damage after Stroke
2.2. Overexpression of ERK2wt Decreases Blood–Brain Barrier Stability
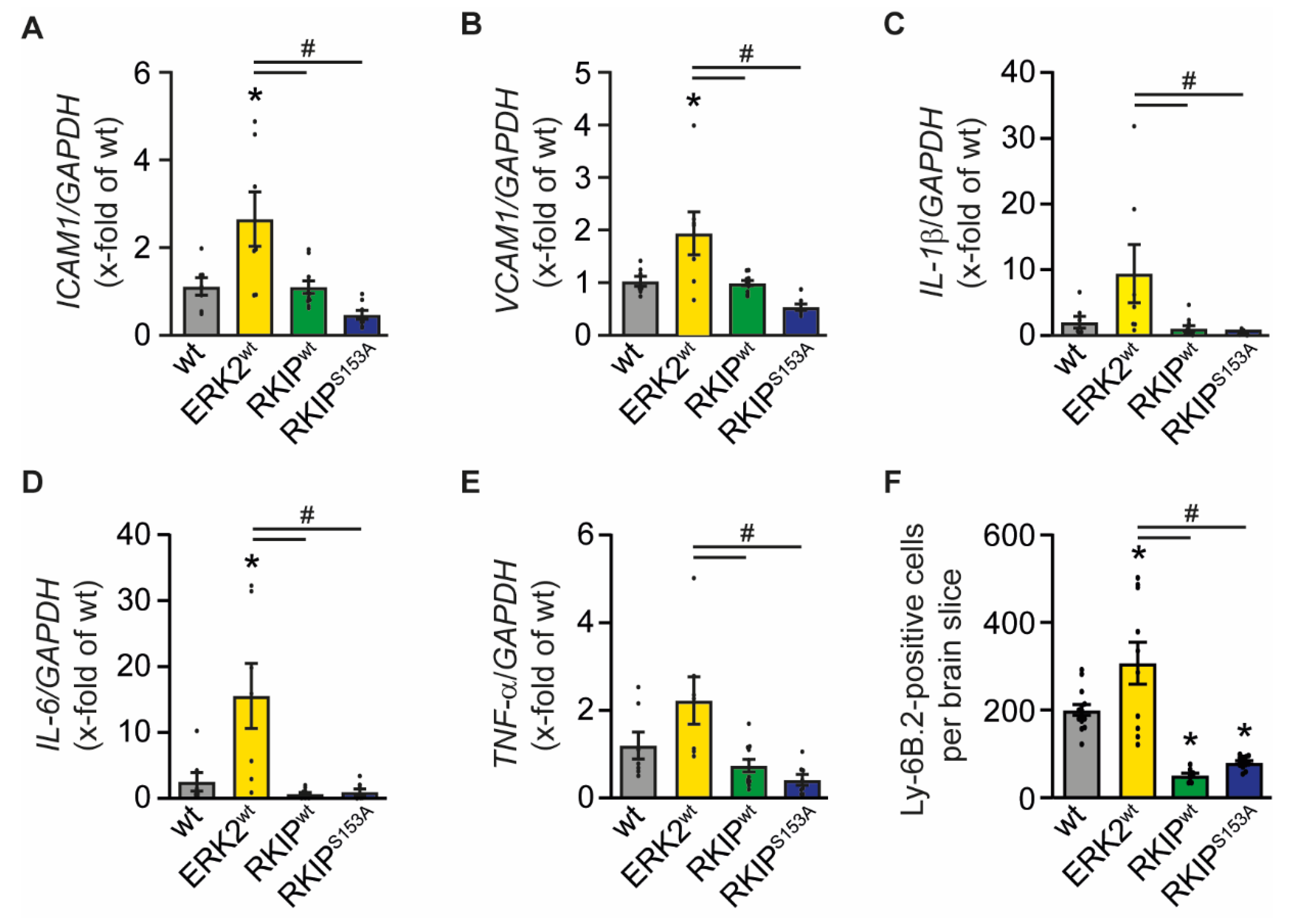
2.3. Overexpression of ERK2wt Increases Inflammation after Stroke
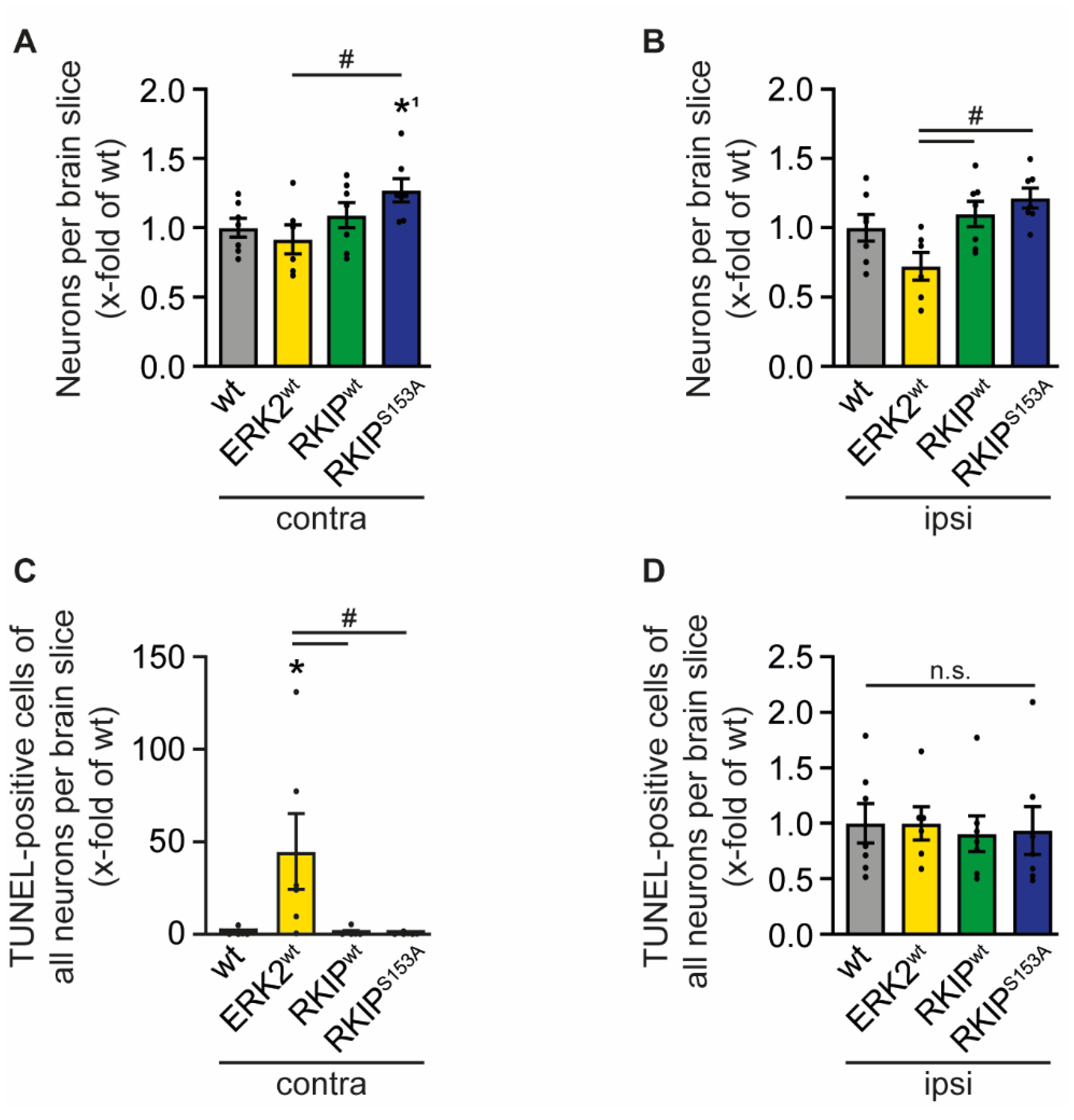
2.4. ERK2wt Overexpression Effects Neuronal Apoptosis
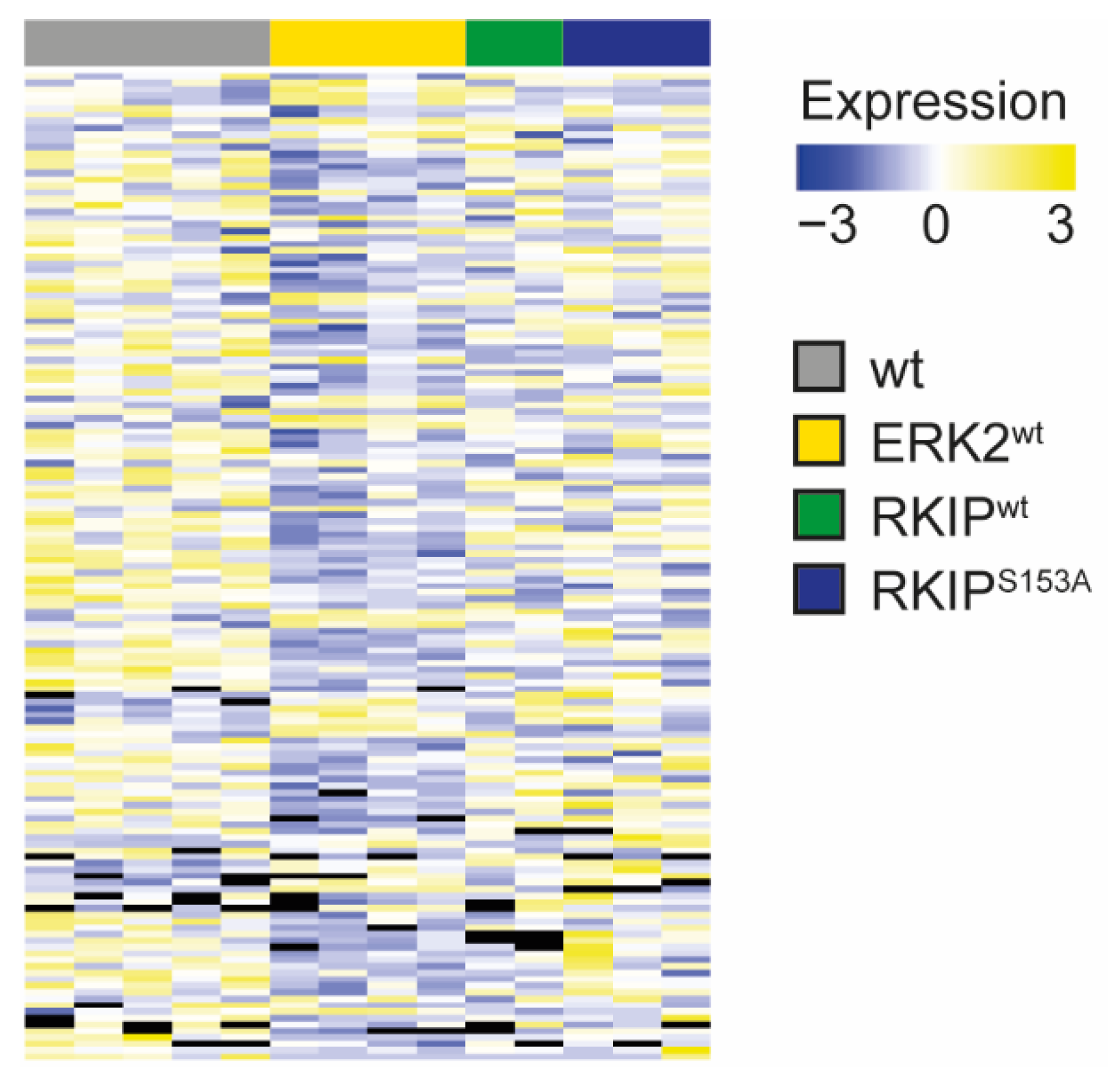
2.5. Proteomic Analyses Revealed Massive Changes in ERK2wt after tMCAO
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Ischemia Model
4.3. Exclusion Criteria
4.4. Assessment of Functional Outcome
4.5. Determination of Infarct Size
4.6. Determination of Blood–Brain Barrier Leakage
4.7. Protein Extraction and Immunoblot Analysis
4.8. RNA Preparation and Quantitative Real-Time PCR
4.9. Immunohistochemistry
4.10. Apoptosis Measurements
4.11. Sample Preparation of Mouse Brain Tissue for Proteomics Analysis
4.12. LC-MS/MS Analysis
4.13. Data Analysis
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Top Ten Causes of Death. 2018. Available online: https://www.who.int/gho/mortality_burden_disease/causes_death/top_10/en/ (accessed on 26 June 2020).
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Ringleb, P.A.; Hamann, G.F.; Röther, J.; Jansen, O.; Groden, C.; Veltkamp, R. Leitlinien für Diagnostik und Therapie in der Neurologie, Akuttherapie des ischämischen Schlaganfalls—Ergänzung 2015, Rekanalisierende Therapie. Aktuelle Neurol. 2016, 43, 82–91. Available online: https://www.dgn.org/images/red_leitlinien/LL_2015/PDFs_Download/030140_LL_akuter-ischaemischer-schlaganfall_final.pdf (accessed on 15 August 2020).
- Veltkamp, R. Leitlinien für Diagnostik und Therapie in der Neurologie, Kapitel: Vaskuläre Erkrankungen, S1 Leitlinie Akuttherapie des Ischämischen Schlaganfalls 2012. Leitlinien Diagnostik Therapie Neurologie 2012, 307–324. Available online: https://www.dsginfo.de/images/stories/DSG/PDF/Leitlinien/LL_22_2012_akuttherapie_des_ischaemischen_schlaganfalls.pdf (accessed on 15 August 2020).
- Kong, T.; Liu, M.; Ji, B.; Bai, B.; Cheng, B.; Wang, C. Role of the Extracellular Signal-Regulated Kinase 1/2 Signaling Pathway in Ischemia-Reperfusion Injury. Front. Physiol. 2019, 10, 1038. [Google Scholar] [CrossRef] [Green Version]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostajeran, M.; Edvinsson, L.; Warfvinge, K.; Singh, R.; Ansar, S. Inhibition of mitogen-activated protein kinase 1/2 in the acute phase of stroke improves long-term neurological outcome and promotes recovery processes in rats. Acta Physiol. 2017, 219, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Gladbach, A.; Van Eersel, J.; Bi, M.; Ke, Y.D.; Ittner, L.M. ERK inhibition with PD184161 mitigates brain damage in a mouse model of stroke. J. Neural Transm. 2014, 121, 543–547. [Google Scholar] [CrossRef]
- Su, L.; Du, H.; Dong, X.; Zhang, X.; Lou, Z. Raf kinase inhibitor protein regulates oxygen-glucose deprivation-induced PC12 cells apoptosis through the NF-kB and ERK pathways. J. Clin. Biochem. Nutr. 2016, 58, 7–15. [Google Scholar] [CrossRef]
- Wang, Z.; Bu, J.; Yao, X.; Liu, C.; Shen, H.; Li, X.; Li, H.; Chen, G. Phosphorylation at S153 as a Functional Switch of Phosphatidylethanolamine Binding Protein 1 in Cerebral Ischemia-Reperfusion Injury in Rats. Front. Mol. Neurosci. 2017, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, X.; Ma, A.; Kang, Y. Rational Application of β—Hydroxybutyrate Attenuates Ischemic Stroke by Suppressing Oxidative Stress and Mitochondrial- Dependent Apoptosis via Activation of the Erk/CREB/eNOS Pathway. ACS Chem. Neurosci. 2021, 12, 1219–1227. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, T.; He, J.; Liao, Q.; Wang, J. Influence of miR-1 on Nerve Cell Apoptosis in Rats with Cerebral Stroke via Regulating ERK Signaling Pathway. Biomed Res. Int. 2021, 2021, 9988534. [Google Scholar] [CrossRef]
- Chen, W.; Feng, J.; Tong, W. Phosphorylation of astrocytic connexin43 by ERK1/2 impairs blood-brain barrier in acute cerebral ischemia. Cell Biosci. 2017, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J. Neuroinflamm. 2010, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S.; Unsicker, K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010, 277, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Zhao, K.; Su, H.; Zhang, P.; Zhao, N. Resveratrol ameliorates brain injury via the TGF-β-mediated ERK signaling pathway in a rat model of cerebral hemorrhage. Exp. Ther. Med. 2019, 18, 3397–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef]
- Schmid, E.; Neef, S.; Berlin, C.; Tomasovic, A.; Kahlert, K.; Nordbeck, P.; Deiss, K.; Denzinger, S.; Herrmann, S.; Wettwer, E.; et al. Cardiac RKIP induces a beneficial β-adrenoceptor-dependent positive inotropy. Nat. Med. 2015, 21, 1298–1306. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Taqi, Z.; Yeung, K.C. RKIP: Much more than Raf Kinase inhibitory protein. J. Cell. Physiol. 2013, 228, 1688–1702. [Google Scholar] [CrossRef]
- Ziv, I.; Fleminger, G.; Djaldetti, R.; Achiron, A.; Melamed, E.; Sokolovsky, M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke 1992, 23, 1014–1016. [Google Scholar] [CrossRef] [Green Version]
- Battey, T.W.K.; Karki, M.; Singhal, A.B.; Wu, O.; Sadaghiani, S.; Campbell, B.C.V.; Davis, S.M.; Donnan, G.A.; Sheth, K.N.; Kimberly, W.T. Brain edema predicts outcome after non-lacunar ischemic stroke. Stroke 2014, 45, 3643–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, P.; Wu, S.; Yi, X.; Wang, C.; Liu, M. Factors associated with favourable outcome in large hemispheric infarctions. BMC Neurol. 2018, 18, 152. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.; Prameya, R.; Dorovini-Zis, K. In Vitro Adhesion and Migration of T Lymphocytes Across Monolayers of Human Brain Microvessel Endothelial Cells: Regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J. Neuropathol. Exp. Neurol. 1999, 58, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, J.; Wang, Y.; Calder, V.L. Lymphocyte adhesion and transendothelial migration in the central nervous system: The role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology 1995, 86, 408–415. [Google Scholar]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor necrosis factor-α expression in ischemic neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–680. [Google Scholar] [CrossRef]
- Smith, C.J.; Emsley, H.C.A.; Gavin, C.M.; Georgiou, R.F.; Vail, A.; Barberan, E.M.; del Zoppo, G.J.; Hallenbeck, J.M.; Rothwell, N.J.; Hopkins, S.J.; et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004, 4, 2. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol.—Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Gu, R.F.; Fang, T.; Nelson, A.; Gyoneva, S.; Gao, B.; Hedde, J.; Henry, K.; Peterson, E.; Burkly, L.C.; Wei, R. Proteomic Characterization of the Dynamics of Ischemic Stroke in Mice. J. Proteome Res. 2021, 20, 3689–3700. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Park, S.; Ha, S.; Kwon, J.S.; Khan, M.R.; Kang, B.G.; Dawson, T.M.; Dawson, V.L.; Andrabi, S.A.; Kang, S.U. Quantitative mass spectrometric analysis of the mouse cerebral cortex after ischemic stroke. PLoS ONE 2020, 15, e0231978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Zhou, Y.T.; Zeng, Y.F.; Liu, T.; Yang, Z.Y.; Tang, T.; Luo, J.K.; Wang, Y. Proteomics Analysis of Brain Tissue in a Rat Model of Ischemic Stroke in the Acute Phase. Front. Mol. Neurosci. 2020, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Gotoh, T.; Tsuji, K.; Lo, E.H.; Huang, S.; Feig, L.A. Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J. 2004, 23, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Yu, D.; Tong, Y.; Mao, M. Effect of neuroserpin in a neonatal hypoxic-ischemic injury model ex vivo. Biol. Res. 2012, 45, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Yepes, M.; Sandkvist, M.; Wong, M.K.K.; Coleman, T.A.; Smith, E.; Cohan, S.L.; Lawrence, D.A. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood 2000, 96, 569–576. [Google Scholar] [CrossRef]
- Wang, Y.C.; Li, X.; Shen, Y.; Lyu, J.; Sheng, H.; Paschen, W.; Yang, W. PERK (Protein Kinase RNA-Like ER Kinase) Branch of the Unfolded Protein Response Confers Neuroprotection in Ischemic Stroke by Suppressing Protein Synthesis. Stroke 2020, 51, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.W.; Lai, J.H.; Chung-Che Wu, J.; Tsai, Y.-R.; Chen, Y.-H.; Kang, S.-J.; Chiang, Y.-H.; Chang, C.-F.; Chen, K.-Y. Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 2899. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Ding, L.; Su, Y.; Shao, R.; Liu, J.; Huang, Y. Neuroprotective effects of curcumin against rats with focal cerebral ischemia-reperfusion injury. Int. J. Mol. Med. 2019, 43, 1879–1887. [Google Scholar] [CrossRef]
- Breitenbach, T.; Lorenz, K.; Dandekar, T. How to steer and control ERK and the ERK signaling cascade exemplified by looking at cardiac insufficiency. Int. J. Mol. Sci. 2019, 20, 2179. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Zhang, R.; Chen, Y.; Ma, C.; Zhu, Z. Raf Kinase Inhibitor Protein Attenuates Ischemic-Induced Microglia Cell Apoptosis and Activation Through NF-κB Pathway. Cell. Physiol. Biochem. 2017, 41, 1125–1134. [Google Scholar] [CrossRef]
- Jung, H.Y.; Cho, S.B.; Kim, W.; Yoo, D.Y.; Won, M.-H.; Choi, G.-M.; Cho, T.-G.; Kim, D.W.; Hwang, I.K.; Choi, S.Y.; et al. Phosphatidylethanolamine-binding protein 1 protects CA1 neurons against ischemic damage via ERK-CREB signaling in Mongolian gerbils. Neurochem. Int. 2018, 118, 265–274. [Google Scholar] [CrossRef]
- Yang, X.; Asakawa, T.; Han, S.; Liu, L.; Li, W.; Wu, W.; Luo, Y.; Cao, W.; Cheng, X.; Xiao, B.; et al. Neuroserpin protects rat neurons and microglia-mediated inflammatory response against oxygen-glucose deprivation- and reoxygenation treatments in an in vitro study. Cell. Physiol. Biochem. 2016, 38, 1472–1482. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, S.; Chen, Y.; Li, L.; Li, X.; Qu, Z.; Huang, J.; Fan, L.; Yuan, C.; Song, N.; et al. Smoothened is a therapeutic target for reducing glutamate toxicity in ischemic stroke. Sci. Transl. Med. 2021, 13, eaba3444. [Google Scholar] [CrossRef]
- Tomasovic, A.; Brand, T.; Schanbacher, C.; Kramer, S.; Hümmert, M.W.; Godoy, P.; Schmidt-Heck, W.; Nordbeck, P.; Ludwig, J.; Homann, S.; et al. Interference with ERK-dimerization at the nucleocytosolic interface targets pathological ERK1/2 signaling without cardiotoxic side-effects. Nat. Commun. 2020, 11, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.A.; Benedek, G.; Liang, J.; Nguyen, H.; Kent, G.; Vandenbark, A.A.; Saugstad, J.A.; Offner, H. Sex Differences in Regulatory Cells in Experimental Stroke. Cell Immunol. 2017, 318, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Bieber, M.; Schuhmann, M.K.; Volz, J.; Kumar, G.J.; Vaidya, J.R.; Nieswandt, B.; Pham, M.; Stoll, G.; Kleinschnitz, C.; Kraft, P. Description of a Novel Phosphodiesterase (PDE)-3 Inhibitor Protecting Mice from Ischemic Stroke Independent from Platelet Function. Stroke 2019, 50, 478–486. [Google Scholar] [CrossRef]
- Bederson, J.B.; Pitts, L.H.; Tsuji, M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986, 17, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Langhauser, F.; Kraft, P.; Göb, E.; Leinweber, J.; Schuhmann, M.K.; Lorenz, K.; Gelderblom, M.; Bittner, S.; Meuth, S.G.; Wiendl, H.; et al. Blocking of α4 integrin does not protect from acute ischemic stroke in mice. Stroke 2014, 45, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Kraft, P.; Schuhmann, M.K.; Fluri, F.; Lorenz, K.; Zernecke, A.; Stoll, G.; Nieswandt, B.; Kleinschnitz, C. Efficacy and safety of platelet glycoprotein receptor blockade in aged and comorbid mice with acute experimental stroke. Stroke 2015, 46, 3502–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, M.K.; Guthmann, J.; Stoll, G.; Nieswandt, B.; Kraft, P.; Kleinschnitz, C. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J. Neuroinflamm. 2017, 14, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, C.; Deiss, K.; Herrmann, S.; Vidal, M.; Oezkur, M.; Gorski, A.; Weidemann, F.; Lohse, M.J.; Lorenz, K. Interference with ERKThr188 phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 7440–7445. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hentschel, A.; Czech, A.; Münchberg, U.; Freier, E.; Schara-Schmidt, U.; Sickmann, A.; Reimann, J.; Roos, A. Protein signature of human skin fibroblasts allows the study of the molecular etiology of rare neurological diseases. Orphanet J. Rare Dis. 2021, 16, 73. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Schumbrutzki, C.; Wortelkamp, S.; Sickmann, A.; Zahedi, R.P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteom. 2012, 75, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.D.L.; Malchow, S.; Reich, S.; Steltgens, S.; Shuvaev, K.V.; Loroch, S.; Lorenz, C.; Sickmann, A.; Knobbe-Thomsen, C.B.; Tews, B.; et al. A sensitive and simple targeted proteomics approach to quantify transcription factor and membrane proteins of the unfolded protein response pathway in glioblastoma cells. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Olsen, J.V.; de Godoy, L.M.F.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, Y.; Bateman, N.W.; Gallien, S.; Goetze, S.; Zhou, Y.; Navarro, P.; Hu, M.; Parikh, N.; Hood, B.L.; Conrads, K.A.; et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Perez-riverol, Y.; Csordas, A.; Bai, J.; Bernal-llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schanbacher, C.; Bieber, M.; Reinders, Y.; Cherpokova, D.; Teichert, C.; Nieswandt, B.; Sickmann, A.; Kleinschnitz, C.; Langhauser, F.; Lorenz, K. ERK1/2 Activity Is Critical for the Outcome of Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 706. https://doi.org/10.3390/ijms23020706
Schanbacher C, Bieber M, Reinders Y, Cherpokova D, Teichert C, Nieswandt B, Sickmann A, Kleinschnitz C, Langhauser F, Lorenz K. ERK1/2 Activity Is Critical for the Outcome of Ischemic Stroke. International Journal of Molecular Sciences. 2022; 23(2):706. https://doi.org/10.3390/ijms23020706
Chicago/Turabian StyleSchanbacher, Constanze, Michael Bieber, Yvonne Reinders, Deya Cherpokova, Christina Teichert, Bernhard Nieswandt, Albert Sickmann, Christoph Kleinschnitz, Friederike Langhauser, and Kristina Lorenz. 2022. "ERK1/2 Activity Is Critical for the Outcome of Ischemic Stroke" International Journal of Molecular Sciences 23, no. 2: 706. https://doi.org/10.3390/ijms23020706