
“Bioinspired” Membrane-Coated Nanosystems in Cancer Theranostics: A Comprehensive Review

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Membrane Sources and Attributes
2.1. Blood Cells
- Macrophage: C-C chemokine receptor 2 (CCR2), vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1) facilitate the movement towards inflammatory tumor sites. α4 and β1 integrins interact with VCAM-1 on cancer cell membranes, allowing selective interaction with target cancer cells [31]. In addition, CD45, CD11a, and glycans act as functional molecules that aid in tumor localization by preventing internalization by phagocytes [32].
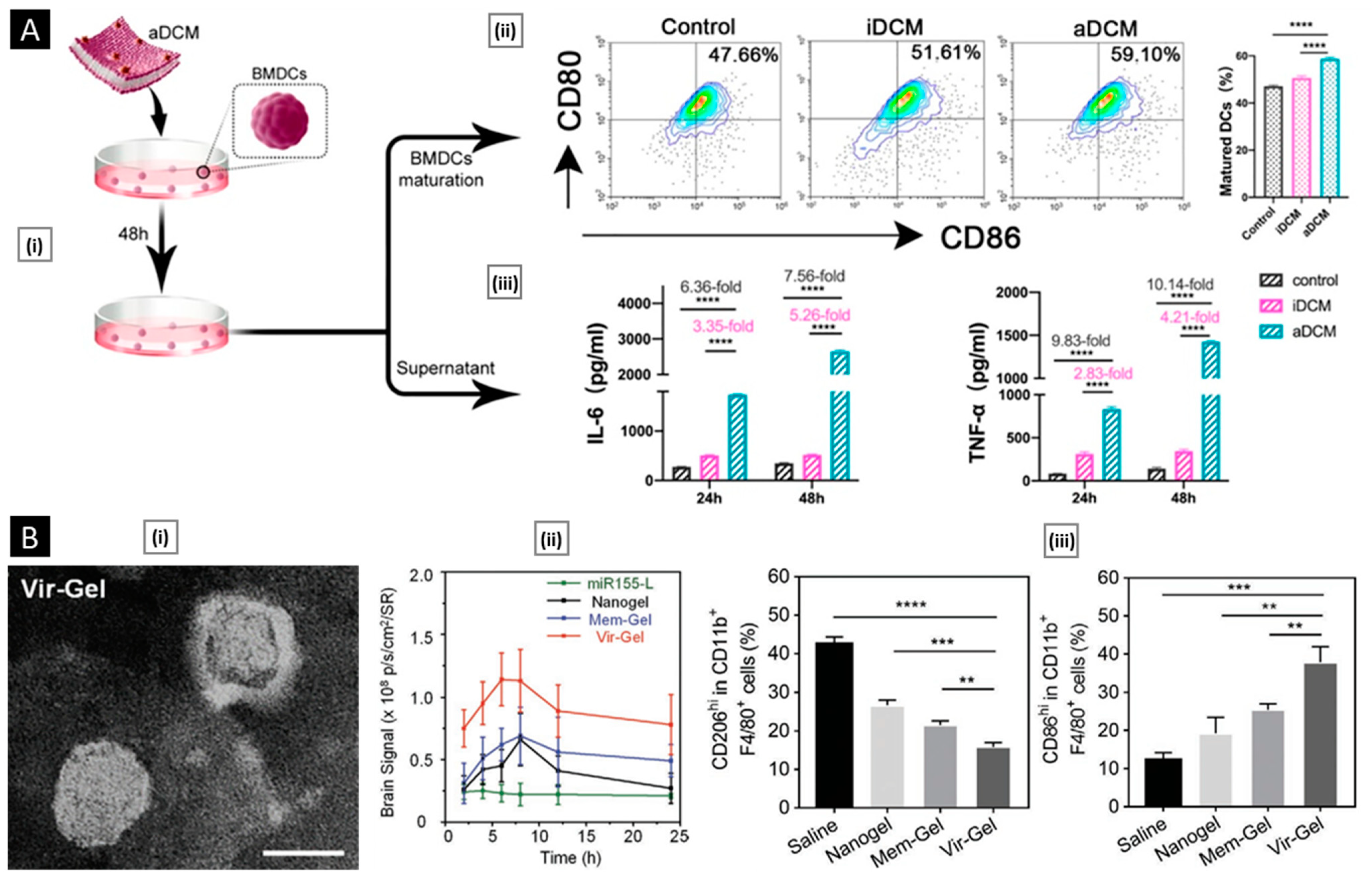
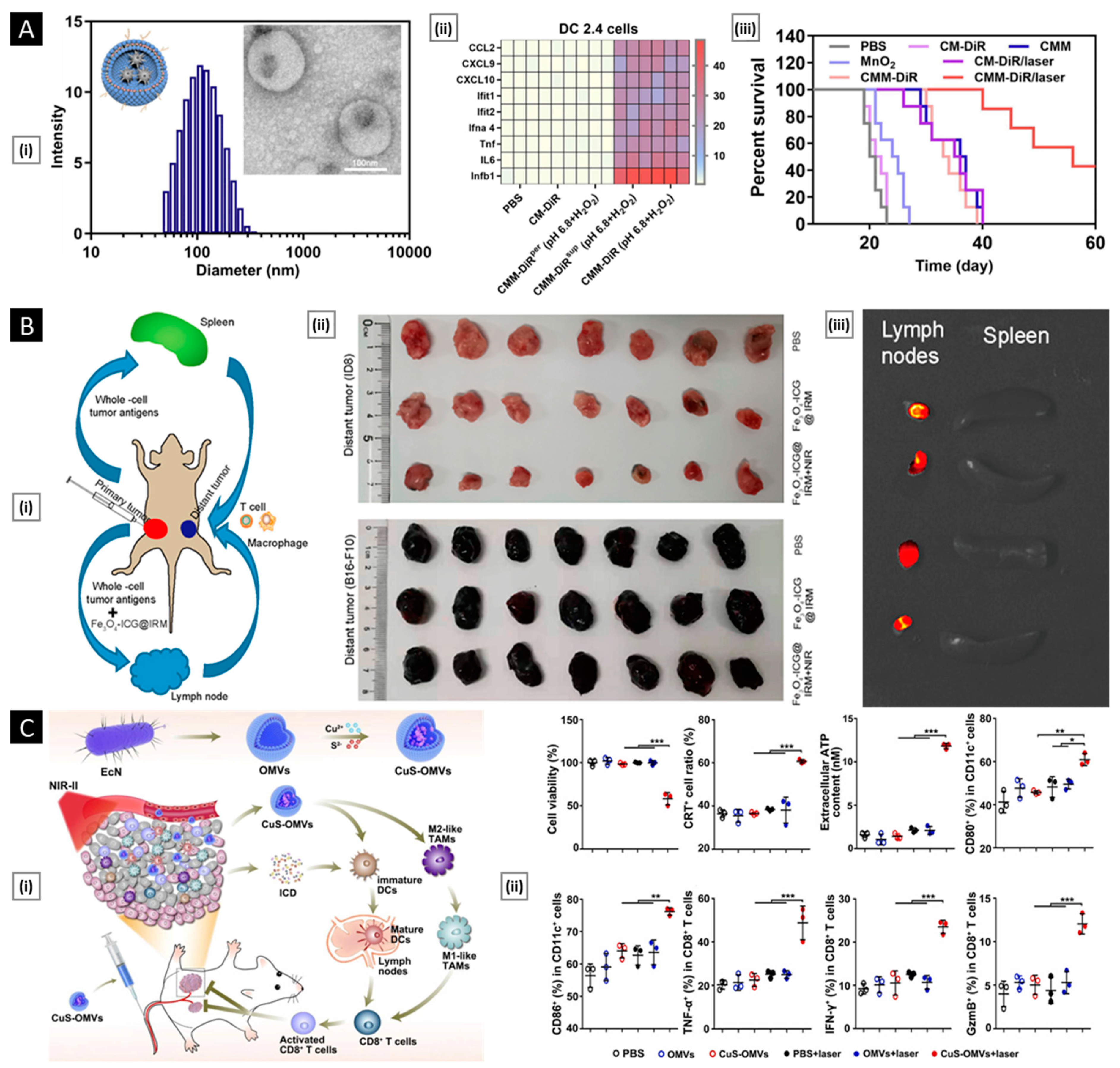
- DCs: They activate T cells by presenting antigens through their broad spectrum of membrane peptide/MHC complexes. ICAM-3, CD40, CD44, and integrins are a few molecules that aid in the adhesion and interaction of DCs [33]. DCs, as a membrane source, provide the advantage of lymph node targeting via the CCR7 receptor [34].
- NK cells: They play a crucial role in cancer elimination by monitoring the atypical expression of MHC-I and stress proteins on the cell surface. Despite the absence of tumor antigen-specific cell surface receptors, membranes sourced from NK cells possess several alternative receptors (such as NKG2D, NKp44, NKp46, NKp30, and DNAM-1) that enable them to recognize cancer cells, enhancing biocompatibility and tumor homing ability [35,36].
2.2. Cancer Cells
2.3. Stem Cells
2.4. Extracellular Vesicles
2.5. Viral Capsids
2.6. Bacteria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Source | Surface Markers/ Proteins | Key Advantages | Limitations | Ref. |
|---|---|---|---|---|
| Erythrocytes | CD47, C8 binding protein | Prolonged circulation time; ease of isolation; reduced susceptibility to macrophage uptake | Absence of tumor-specific ligands | [20] |
| Platelets | CD62p, PECAM-1, CD44, CD47 | Prolonged circulation time; ease of isolation; robust immune evasion | Aggregation of coated nanoparticles; absence of tumor-specific ligands | [26] |
| Macrophages | CCR2, VCAM-1, ICAM-1 | Facilitates immune cell trafficking towards tumor; evades reticuloendothelial system; trans-endothelial migration through intercellular adhesion | Limited tumor-targeting ability | [32] |
| Dendritic cells | Peptide/MHC Complex, ICAM-3, CD40, CD44, CCR7 | Upregulated co-stimulatory molecules; antigen-specific T-cell activation | Poor sensitivity and specificity of peptide/MHC complex to bind to CD8+ cells | [89] |
| Natural killer cells | NKG2D, NKp44, NKp46, NKp30, DNAM-1 | Tumor recognition, a wide range of tumor targeting | Restricted proliferation of primary NK cells; lower infiltration in solid tumors | [90] |
| Cancer cells | CCAM, CD44, IG-SF | Strong adhesion among homotypic tumor cells, source of tumor antigens | Laborious isolation process; cell culture conditions, passage number, and genetic drift can induce variability in surface markers | [42] |
| Mesenchymal stem cells | CXCR4, PDGFR, VEGFR, E-selectin, P-selectin | Natural affinity toward tumor cells | Varying composition of the cell membrane may lead to ineffective therapeutic response | [48] |
| Exosomes | CD9, CD63, Alix, EP-CAM, CD55, CD59, MHC | Low immunogenicity; efficient cellular uptake; intrinsic tumor targeting | Laborious isolation process; presence of inherent biological cargo can cause unwanted biological effects | [57] |
| Viral Capsids | AAV-Rep78, Parvovirus NS1 | Activates host immune system; selective apoptosis of tumor cells | Non-specific binding to healthy cells may lead to immunogenic responses | [65] |
| Bacteria | OmpA, OmpC, AcrA | Immune cell activation; self-adjuvant characteristic | Pathogenicity needs to be adequately addressed before in vivo use | [82] |
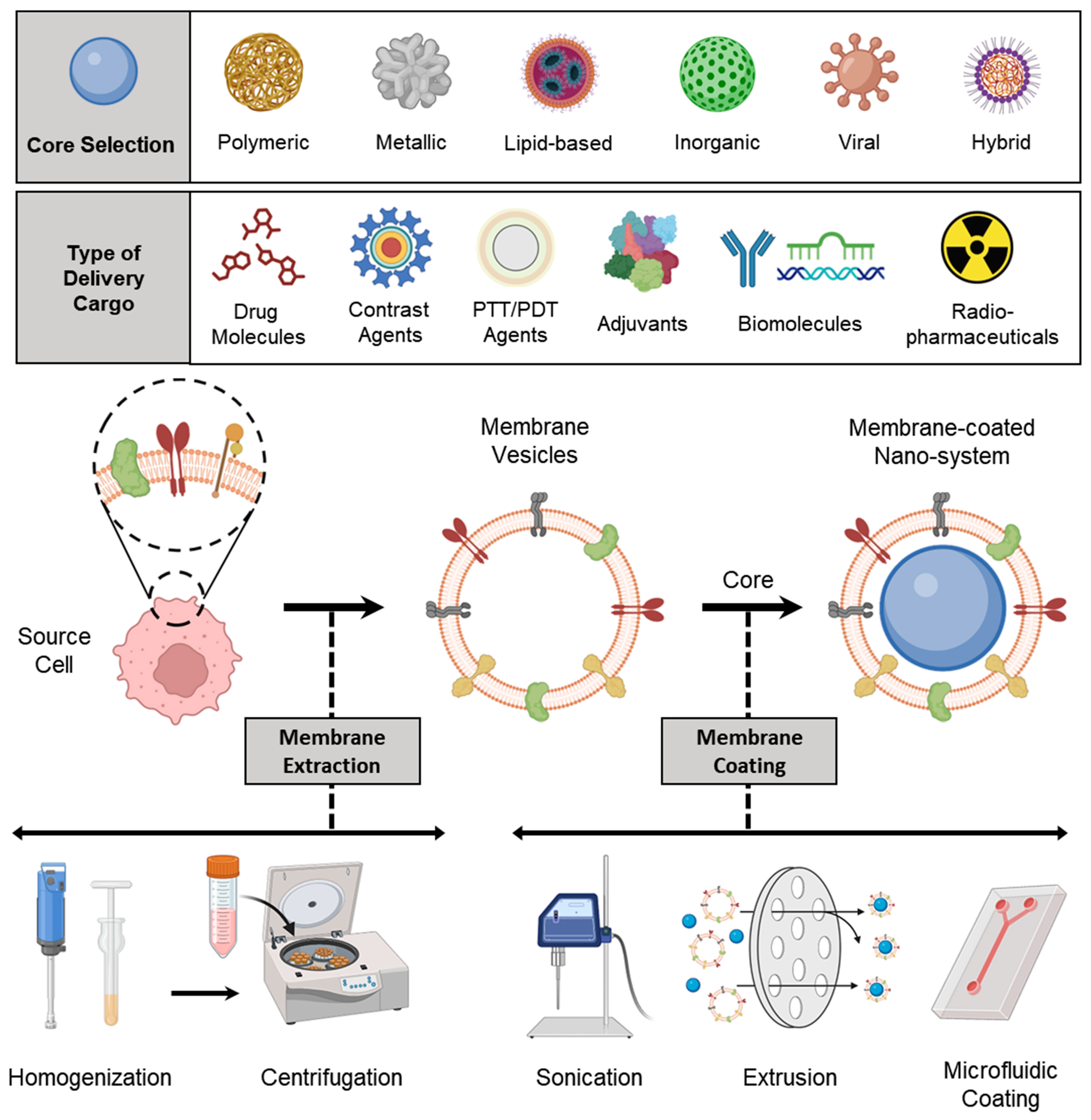
3. Preparation of Membrane-Coated Nanosystems
3.1. Membrane Isolation/Extraction
3.2. Selection of NP Core
3.3. Membrane Coating
3.3.1. Physical Extrusion
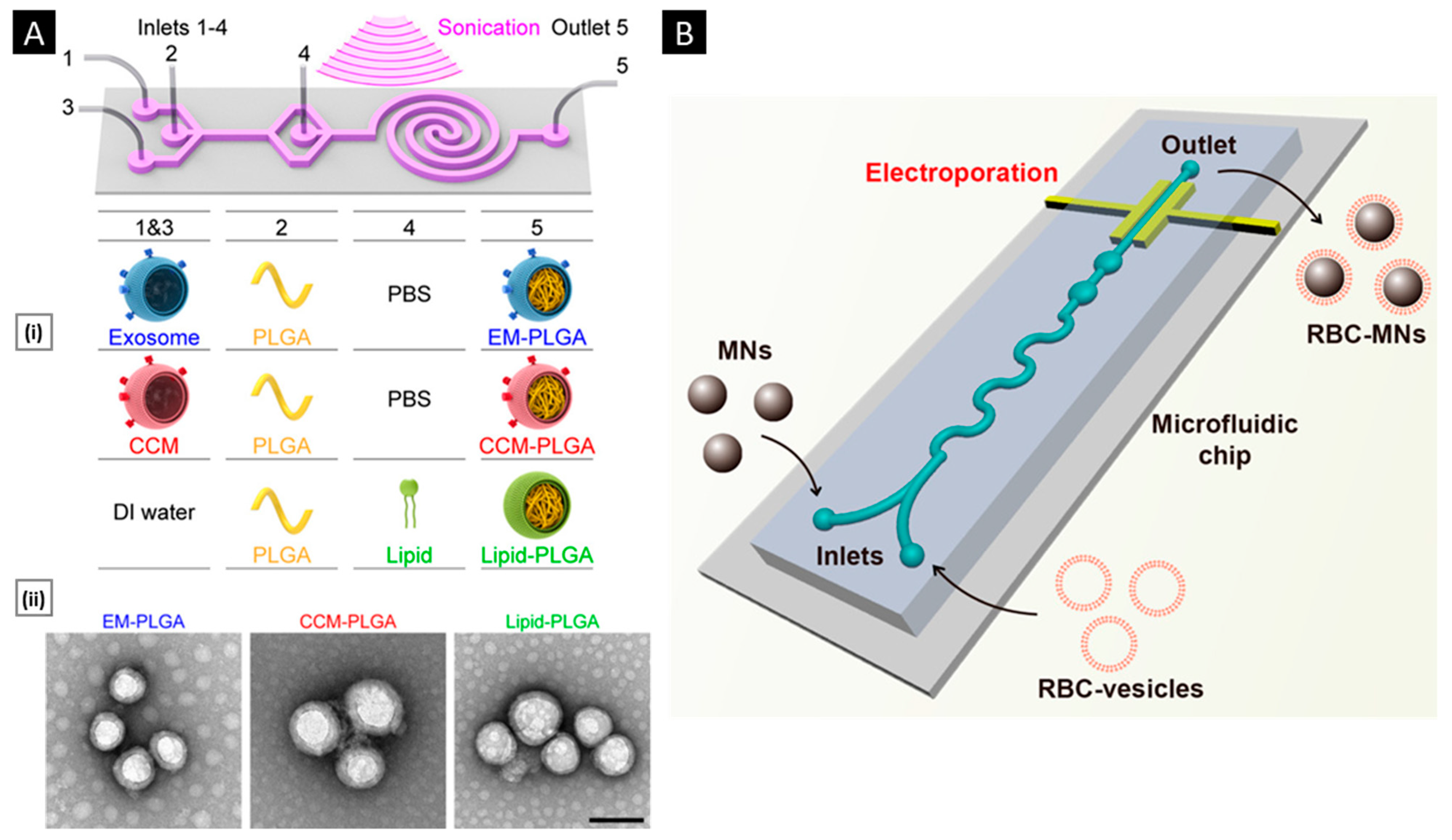
3.3.2. Sonication
3.3.3. Microfluidic Coating
3.4. Other Special Approaches
4. Methods of Characterization
4.1. Physicochemical Properties
4.1.1. Size and Surface Attributes
4.1.2. Membrane Properties

4.2. Biological Properties
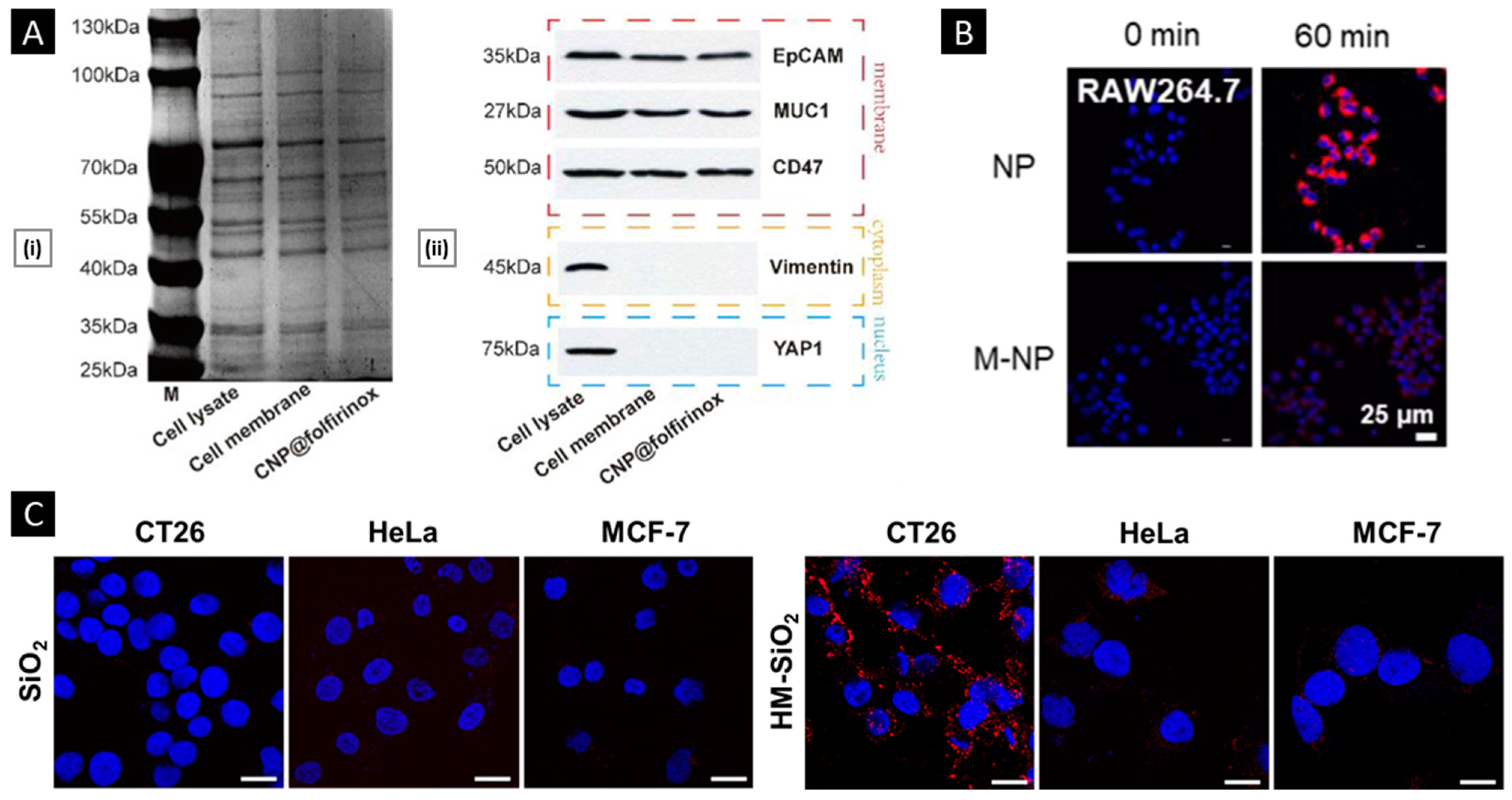
4.2.1. Verifications of Membrane Proteins
4.2.2. In Vitro Functional Validation
5. Functionalization Approaches
5.1. Lipid Insertion
5.2. Hybridization
5.3. Metabolic Engineering

5.4. Genetic Engineering
6. Theranostics Applications
6.1. Tumor Diagnosis and Bio-Imaging


6.2. Anti-Tumor Therapy


7. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salave, S.; Rana, D.; Pardhe, R.; Bule, P.; Benival, D. Unravelling Micro and Nano Vesicular System in Intranasal Drug Delivery for Epilepsy. Pharm. Nanotechnol. 2022, 10, 182–193. [Google Scholar]
- Rana, D.; Salave, S.; Longare, S.; Agarwal, R.; Kalia, K.; Benival, D. Nanotherapeutics in Tumour Microenvironment for Cancer Therapy. Nanosci. Nanotechnol. Asia 2022, 12, 32–47. [Google Scholar] [CrossRef]
- Palekar-Shanbhag, P.; Jog, S.V.; Chogale, M.M.; Gaikwad, S.S. Theranostics for Cancer Therapy. Curr. Drug Deliv. 2013, 10, 357–362. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An Overview of Active and Passive Targeting Strategies to Improve the Nanocarriers Efficiency to Tumour Sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Onzi, G.; Guterres, S.S.; Pohlmann, A.R.; Frank, L.A. Passive Targeting and the Enhanced Permeability and Retention (EPR) Effect. In The ADME Encyclopedia; Springer: Cham, Switzerland, 2021; pp. 1–13. [Google Scholar]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Zhou, Q.; Dong, C.; Fan, W.; Jiang, H.; Xiang, J.; Qiu, N.; Piao, Y.; Xie, T.; Luo, Y.; Li, Z.; et al. Tumor Extravasation and Infiltration as Barriers of Nanomedicine for High Efficacy: The Current Status and Transcytosis Strategy. Biomaterials 2020, 240, 119902. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, F.; Mishra, R.K.; Khan, R. Precision Cancer Nanotherapy: Evolving Role of Multifunctional Nanoparticles for Cancer Active Targeting. J. Med. Chem. 2019, 62, 10475–10496. [Google Scholar] [CrossRef]
- Raj, S.; Khurana, S.; Choudhari, R.; Kesari, K.K.; Kamal, M.A.; Garg, N.; Ruokolainen, J.; Das, B.C.; Kumar, D. Specific Targeting Cancer Cells with Nanoparticles and Drug Delivery in Cancer Therapy. Semin. Cancer Biol. 2021, 69, 166–177. [Google Scholar] [CrossRef]
- Liu, H.; Su, Y.Y.; Jiang, X.C.; Gao, J.Q. Cell Membrane-Coated Nanoparticles: A Novel Multifunctional Biomimetic Drug Delivery System. Drug Deliv. Transl. Res. 2023, 13, 716–737. [Google Scholar] [CrossRef]
- Pasto, A.; Giordano, F.; Evangelopoulos, M.; Amadori, A.; Tasciotti, E. Cell Membrane Protein Functionalization of Nanoparticles as a New Tumor-Targeting Strategy. Clin. Transl. Med. 2019, 8, 8. [Google Scholar] [CrossRef]
- Cai, R.; Chen, C. The Crown and the Scepter: Roles of the Protein Corona in Nanomedicine. Adv. Mater. 2019, 31, 201805740. [Google Scholar] [CrossRef]
- Lei, W.; Yang, C.; Wu, Y.; Ru, G.; He, X.; Tong, X.; Wang, S. Nanocarriers Surface Engineered with Cell Membranes for Cancer Targeted Chemotherapy. J. Nanobiotechnol. 2022, 20, 45. [Google Scholar] [CrossRef]
- Polaka, S.; Desai, N.; Kshirsagar, B.; Rajpoot, K.; Tekade, R.K. Revamping the Pharmacokinetics of Poorly Soluble Drugs Using Different Formulations. In Biopharmaceutics and Pharmacokinetics Considerations; Academic Press: Cambridge, MA, USA, 2021; pp. 387–413. [Google Scholar]
- Qie, Y.; Yuan, H.; Von Roemeling, C.A.; Chen, Y.; Liu, X.; Shih, K.D.; Knight, J.A.; Tun, H.W.; Wharen, R.E.; Jiang, W.; et al. Surface Modification of Nanoparticles Enables Selective Evasion of Phagocytic Clearance by Distinct Macrophage Phenotypes. Sci. Rep. 2016, 6, 26269. [Google Scholar] [CrossRef] [Green Version]
- Rana, D.; Salave, S.; Patel, R.; Khunt, D.; Misra, M.; Prajapati, B.; Patel, G.; Patel, J. Solid Lipid Nanoparticles in Tuberculosis. Tubercular Drug Delivery Systems; Springer: Cham, Swizerland, 2023; pp. 99–121. [Google Scholar] [CrossRef]
- Salave, S.; Shinde, S.D.; Rana, D.; Sahu, B.; Kumar, H.; Patel, R.; Benival, D.; Kommineni, N. Peptide Engraftment on PEGylated Nanoliposomes for Bone Specific Delivery of PTH (1-34) in Osteoporosis. Pharmaceutics 2023, 15, 608. [Google Scholar] [CrossRef]
- Rossi, L.; Pierigè, F.; Antonelli, A.; Bigini, N.; Gabucci, C.; Peiretti, E.; Magnani, M. Engineering Erythrocytes for the Modulation of Drugs’ and Contrasting Agents’ Pharmacokinetics and Biodistribution. Adv. Drug Deliv. Rev. 2016, 106, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.J.; Fang, R.H.; Zhang, L. Erythrocyte-Inspired Delivery Systems. Adv. Healthc. Mater. 2012, 1, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red Blood Cell Membrane-Camouflaged Nanoparticles: A Novel Drug Delivery System for Antitumor Application. Acta Pharm. Sin. B 2019, 9, 675–689. [Google Scholar] [CrossRef]
- Hu, C.M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, S.; Dumoga, S.; Singh, N. Red Blood Cells Membrane-Derived Nanoparticles: Applications and Key Challenges in Their Clinical Translation. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Duan, Y.; Zhang, Q.; Komarla, A.; Gong, H.; Gao, W.; Zhang, L. Drug Targeting via Platelet Membrane-Coated Nanoparticles. Small Struct. 2020, 1, 2000018. [Google Scholar] [CrossRef]
- Gaertner, F.; Massberg, S. Patrolling the Vascular Borders: Platelets in Immunity to Infection and Cancer. Nat. Rev. Immunol. 2019, 19, 747–760. [Google Scholar] [CrossRef]
- Kunde, S.S.; Wairkar, S. Platelet Membrane Camouflaged Nanoparticles: Biomimetic Architecture for Targeted Therapy. Int. J. Pharm. 2021, 598, 120395. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, K.; Zhang, X.; Ouyang, B.; Liu, H.; Pang, Z.; Yang, W. Platelet Membrane-Camouflaged Magnetic Nanoparticles for Ferroptosis-Enhanced Cancer Immunotherapy. Small 2020, 16, 202001704. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Leukocyte-Endothelial-Cell Interactions in Leukocyte Transmigration and the Inflammatory Response. Trends Immunol. 2003, 24, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Oroojalian, F.; Beygi, M.; Baradaran, B.; Mokhtarzadeh, A.; Shahbazi, M.A. Immune Cell Membrane-Coated Biomimetic Nanoparticles for Targeted Cancer Therapy. Small 2021, 17, e2006484. [Google Scholar] [CrossRef]
- Bernardini, G.; Zabel, B.A. Editorial: The Role of Chemoattractants in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2671. [Google Scholar] [CrossRef]
- Cao, H.; Dan, Z.; He, X.; Zhang, Z.; Yu, H.; Yin, Q.; Li, Y. Liposomes Coated with Isolated Macrophage Membrane Can Target Lung Metastasis of Breast Cancer. ACS Nano 2016, 10, 7738–7748. [Google Scholar] [CrossRef]
- Gong, P.; Wang, Y.; Zhang, P.; Yang, Z.; Deng, W.; Sun, Z.; Yang, M.; Li, X.; Ma, G.; Deng, G.; et al. Immunocyte Membrane-Coated Nanoparticles for Cancer Immunotherapy. Cancers 2020, 13, 77. [Google Scholar] [CrossRef]
- Cheng, S.; Xu, C.; Jin, Y.; Li, Y.; Zhong, C.; Ma, J.; Yang, J.; Zhang, N.; Li, Y.; Wang, C.; et al. Artificial Mini Dendritic Cells Boost T Cell-Based Immunotherapy for Ovarian Cancer. Adv. Sci. 2020, 7, 1903301. [Google Scholar] [CrossRef]
- Schineis, P.; Kotkowska, Z.K.; Vogel-Kindgen, S.; Friess, M.C.; Theisen, M.; Schwyter, D.; Hausammann, L.; Subedi, S.; Varypataki, E.M.; Waeckerle-Men, Y.; et al. Photochemical Internalization (PCI)-Mediated Activation of CD8 T Cells Involves Antigen Uptake and CCR7-Mediated Transport by Migratory Dendritic Cells to Draining Lymph Nodes. J. Control. Release 2021, 332, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Sun, Z.; Li, S.; Peng, X.; Li, W.; Zhou, L.; Ma, Y.; Gong, P.; Cai, L. Cell-Membrane Immunotherapy Based on Natural Killer Cell Membrane Coated Nanoparticles for the Effective Inhibition of Primary and Abscopal Tumor Growth. ACS Nano 2018, 12, 12096–12108. [Google Scholar] [CrossRef] [PubMed]
- Farcas, M.; Inngjerdingen, M. Natural Killer Cell-Derived Extracellular Vesicles in Cancer Therapy. Scand. J. Immunol. 2020, 92, e12938. [Google Scholar] [CrossRef]
- Rana, D.; Salave, S.; Perla, A.; Nadkarni, A.; Kolhe, S.; Jindal, A.B.; Mandoli, A.; Dwivedi, P.; Benival, D. Bugs as Drugs: Understanding the Linkage between Gut Microbiota and Cancer Treatment. Curr. Drug Targets 2022, 23, 869–888. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jones, R.; Liu, J.C.; Deng, T.; Robinson, T.; Chung, P.E.D.; Wang, S.; Herschkowitz, J.I.; Egan, S.E.; Perou, C.M.; et al. RB1 and P53 at the Crossroad of EMT and Triple-Negative Breast Cancer. Cell Cycle 2011, 10, 1563–1570. [Google Scholar] [CrossRef]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef] [Green Version]
- Lian, S.; Xie, X.; Lu, Y.; Jia, L. Checkpoint CD47 Function On Tumor Metastasis And Immune Therapy. OncoTargets Ther. 2019, 12, 9105–9114. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell Adhesion in Cancer: Beyond the Migration of Single Cells. J. Biol. Chem. 2020, 295, 2495. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Dong, X.W.; Guo, X.L. Role of the Interaction between Galectin-3 and Cell Adhesion Molecules in Cancer Metastasis. Biomed. Pharmacother. 2015, 69, 179–185. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.M.J.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef]
- Rao, L.; Yu, G.T.; Meng, Q.F.; Bu, L.L.; Tian, R.; Lin, L.S.; Deng, H.; Yang, W.; Zan, M.; Ding, J.; et al. Cancer Cell Membrane-Coated Nanoparticles for Personalized Therapy in Patient-Derived Xenograft Models. Adv. Funct. Mater. 2019, 29, 1905671. [Google Scholar] [CrossRef]
- Wang, M.; Xin, Y.; Cao, H.; Li, W.; Hua, Y.; Webster, T.J.; Zhang, C.; Tang, W.; Liu, Z. Recent Advances in Mesenchymal Stem Cell Membrane-Coated Nanoparticles for Enhanced Drug Delivery. Biomater. Sci. 2021, 9, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Giuliani, M. Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment. Vaccines 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibacher, J.; Henschler, R. Biodistribution, Migration and Homing of Systemically Applied Mesenchymal Stem/Stromal Cells. Stem Cell Res. Ther. 2016, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.H.; Zhou, Y.; Tabata, Y.; Gao, J.Q. Mesenchymal Stem Cell-Based Drug Delivery Strategy: From Cells to Biomimetic. J. Control. Release 2019, 294, 102–113. [Google Scholar] [CrossRef]
- Seo, K.W.; Lee, H.W.; Oh, Y.I.; Ahn, J.O.; Koh, Y.R.; Oh, S.H.; Kang, S.K.; Youn, H.Y. Anti-Tumor Effects of Canine Adipose Tissue-Derived Mesenchymal Stromal Cell-Based Interferon-β Gene Therapy and Cisplatin in a Mouse Melanoma Model. Cytotherapy 2011, 13, 944–955. [Google Scholar] [CrossRef]
- Słomka, A.; Urban, S.K.; Lukacs-Kornek, V.; Żekanowska, E.; Kornek, M. Large Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front. Immunol. 2018, 9, 2723. [Google Scholar] [CrossRef] [Green Version]
- Bazzan, E.; Tinè, M.; Casara, A.; Biondini, D.; Semenzato, U.; Cocconcelli, E.; Balestro, E.; Damin, M.; Radu, C.M.; Turato, G.; et al. Critical Review of the Evolution of Extracellular Vesicles’ Knowledge: From 1946 to Today. Int. J. Mol. Sci. 2021, 22, 6417. [Google Scholar] [CrossRef]
- Desai, N.; Gadeval, A.; Kathar, U.; Sengupta, P.; Kalia, K.; Tekade, R.K. Emerging Roles and Biopharmaceutical Applications of Milk Derived Exosomes. J. Drug Deliv. Sci. Technol. 2021, 64, 102577. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane Vesicles, Current State-of-the-Art: Emerging Role of Extracellular Vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [Green Version]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding Microvesicles: Artefacts No More. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, N.; Pishavar, E.; Baradaran, B.; Oroojalian, F.; Mokhtarzadeh, A. Stem Cell Membrane, Stem Cell-Derived Exosomes and Hybrid Stem Cell Camouflaged Nanoparticles: A Promising Biomimetic Nanoplatforms for Cancer Theranostics. J. Control. Release 2022, 348, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Broecker, F. Viruses and Evolution—Viruses First? A Personal Perspective. Front. Microbiol. 2019, 10, 523. [Google Scholar] [CrossRef] [Green Version]
- Salave, S.; Rana, D.; Bodar, A.; Khunt, D.; Prajapati, B.; Patel, J. Viral Infections: Current Treatment Options. In Viral Drug Delivery Systems; Springer: Cham, Switzerland, 2023; pp. 65–89. [Google Scholar]
- Karunakaran, B.; Gupta, R.; Patel, P.; Salave, S.; Sharma, A.; Desai, D.; Benival, D.; Kommineni, N. Emerging Trends in Lipid-Based Vaccine Delivery: A Special Focus on Developmental Strategies, Fabrication Methods, and Applications. Vaccines 2023, 11, 661. [Google Scholar] [CrossRef]
- Fenner, F.; Bachmann, P.A.; Gibbs, E.P.J.; Murphy, F.A.; Studdert, M.J.; White, D.O. Structure and Composition of Viruses. Vet. Virol. 1987, 3–19. [Google Scholar] [CrossRef]
- Louten, J. Virus Structure and Classification. Essent. Hum. Virol. 2016, 19–29. [Google Scholar] [CrossRef]
- Manocha, E.; Caruso, A.; Caccuri, F. Viral Proteins as Emerging Cancer Therapeutics. Cancers 2021, 13, 2199. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Durfee, P.N.; Buley, M.D.; Lino, C.A.; Padilla, D.P.; Phillips, B.; Carter, M.B.; Willman, C.L.; et al. Cell-Specific Delivery of Diverse Cargos by Bacteriophage MS2 Virus-like Particles. ACS Nano 2011, 5, 5729–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ifon, E.T.; Pang, A.L.Y.; Johnson, W.; Cashman, K.; Zimmerman, S.; Muralidhar, S.; Chan, W.Y.; Casey, J.; Rosenthal, L.J. U94 Alters FN1 and ANGPTL4 Gene Expression and Inhibits Tumorigenesis of Prostate Cancer Cell Line PC3. Cancer Cell Int. 2005, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristov, G.; Krämer, M.; Li, J.; El-Andaloussi, N.; Mora, R.; Daeffler, L.; Zentgraf, H.; Rommelaere, J.; Marchini, A. Through Its Nonstructural Protein NS1, Parvovirus H-1 Induces Apoptosis via Accumulation of Reactive Oxygen Species. J. Virol. 2010, 84, 5909–5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.J.; Kalman-Maltese, V.; Cao, X.; Yang, Q.; Trempe, J.P. Inhibition of Adenovirus Cytotoxicity, Replication, and E2a Gene Expression by Adeno-Associated Virus. Virology 2001, 291, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Alemzadeh, E.; Dehshahri, A.; Dehghanian, A.R.; Afsharifar, A.; Behjatnia, A.A.; Izadpanah, K.; Ahmadi, F. Enhanced Anti-Tumor Efficacy and Reduced Cardiotoxicity of Doxorubicin Delivered in a Novel Plant Virus Nanoparticle. Colloids Surf. B Biointerfaces 2019, 174, 80–86. [Google Scholar] [CrossRef]
- Lewis, J.D.; Destito, G.; Zijlstra, A.; Gonzalez, M.J.; Quigley, J.P.; Manchester, M.; Stuhlmann, H. Viral Nanoparticles as Tools for Intravital Vascular Imaging. Nat. Med. 2006, 12, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Raja, K.S.; Wang, Q.; Gonzalez, M.J.; Manchester, M.; Johnson, J.E.; Finn, M.G. Hybrid Virus-Polymer Materials. 1. Synthesis and Properties of PEG-Decorated Cowpea Mosaic Virus. Biomacromolecules 2003, 4, 472–476. [Google Scholar] [CrossRef]
- Massa, S.; Simeone, P.; Muller, A.; Benvenuto, E.; Venuti, A.; Franconi, R. Antitumor Activity of DNA Vaccines Based on the Human Papillomavirus-16 E7 Protein Genetically Fused to a Plant Virus Coat Protein. Hum. Gene Ther. 2008, 19, 354–364. [Google Scholar] [CrossRef]
- Peacey, M.; Wilson, S.; Perret, R.; Ronchese, F.; Ward, V.K.; Young, V.; Young, S.L.; Baird, M.A. Virus-like Particles from Rabbit Hemorrhagic Disease Virus Can Induce an Anti-Tumor Response. Vaccine 2008, 26, 5334–5337. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Jan, A.T. Outer Membrane Vesicles (OMVs) of Gram-Negative Bacteria: A Perspective Update. Front. Microbiol. 2017, 8, 1053. [Google Scholar] [CrossRef] [PubMed]
- Sartorio, M.G.; Pardue, E.J.; Feldman, M.F.; Haurat, M.F. Bacterial Outer Membrane Vesicles: From Discovery to Applications. Annu. Rev. Microbiol. 2021, 75, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.G.; Work, E. An Extracellular Glycolipid Produced by Escherichia Coli Grown under Lysine-Limiting Conditions. Biochem. J. 1965, 96, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Kulp, A.; Kuehn, M.J. Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecil, J.D.; Sirisaengtaksin, N.; O’Brien-Simpson, N.M.; Krachler, A.M. Outer Membrane Vesicle-Host Cell Interactions. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.H.; Wang, M.Z.; Chu, Y.; Zhang, L.; Niu, J.; Shao, H.T.; Yuan, T.J.; Jiang, Z.H.; Gao, J.Q.; Ning, X.H. Engineering Bacterial Outer Membrane Vesicles as Transdermal Nanoplatforms for Photo-TRAIL-Programmed Therapy against Melanoma. Sci. Adv. 2020, 6, eaba2735. [Google Scholar] [CrossRef]
- Krishnan, N.; Kubiatowicz, L.J.; Holay, M.; Zhou, J.; Fang, R.H.; Zhang, L. Bacterial Membrane Vesicles for Vaccine Applications. Adv. Drug Deliv. Rev. 2022, 185, 114294. [Google Scholar] [CrossRef]
- Gujrati, V.; Kim, S.; Kim, S.H.; Min, J.J.; Choy, H.E.; Kim, S.C.; Jon, S. Bioengineered Bacterial Outer Membrane Vesicles as Cell-Specific Drug-Delivery Vehicles for Cancer Therapy. ACS Nano 2014, 8, 1525–1537. [Google Scholar] [CrossRef]
- Patel, R.B.; Ye, M.; Carlson, P.M.; Jaquish, A.; Zangl, L.; Ma, B.; Wang, Y.; Arthur, I.; Xie, R.; Brown, R.J.; et al. Development of an In Situ Cancer Vaccine via Combinational Radiation and Bacterial-Membrane-Coated Nanoparticles. Adv. Mater. 2019, 31, 1902626. [Google Scholar] [CrossRef]
- Wang, D.; Liu, C.; You, S.; Zhang, K.; Li, M.; Cao, Y.; Wang, C.; Dong, H.; Zhang, X. Bacterial Vesicle-Cancer Cell Hybrid Membrane-Coated Nanoparticles for Tumor Specific Immune Activation and Photothermal Therapy. ACS Appl. Mater. Interfaces 2020, 12, 41138–41147. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, W.J.; Johnston, E.L.; Zavan, L.; Bitto, N.J.; Kaparakis-Liaskos, M. Immunomodulatory Roles and Novel Applications of Bacterial Membrane Vesicles. Mol. Immunol. 2021, 134, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-Positive Bacteria Produce Membrane Vesicles: Proteomics-Based Characterization of Staphylococcus Aureus-Derived Membrane Vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Defourny, K.A.Y.; Smid, E.J.; Abee, T. Gram-Positive Bacterial Extracellular Vesicles and Their Impact on Health and Disease. Front. Microbiol. 2018, 9, 1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cintolo, J.A.; Datta, J.; Mathew, S.J.; Czerniecki, B.J. Dendritic Cell-Based Vaccines: Barriers and Opportunities. Future Oncol. 2012, 8, 1273. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Cristina, M.; Biassoni, R.; Moretta, L. Activating Receptors and Coreceptors Involved in Human Natural Killer Cell-Mediated Cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Bigaj-Józefowska, M.J.; Grześkowiak, B.F. Polymeric Nanoparticles Wrapped in Biological Membranes for Targeted Anticancer Treatment. Eur. Polym. J. 2022, 176, 111427. [Google Scholar] [CrossRef]
- Guo, K.; Xiao, N.; Liu, Y.; Wang, Z.; Tóth, J.; Gyenis, J.; Thakur, V.K.; Oyane, A.; Shubhra, Q.T.H. Engineering Polymer Nanoparticles Using Cell Membrane Coating Technology and Their Application in Cancer Treatments: Opportunities and Challenges. Nano Mater. Sci. 2022, 4, 295–321. [Google Scholar] [CrossRef]
- Wang, F.; Deng, Y.; Yu, L.; Zhou, A.; Wang, J.; Jia, J.; Li, N.; Ding, F.; Lian, W.; Liu, Q.; et al. A Macrophage Membrane-Polymer Hybrid Biomimetic Nanoplatform for Therapeutic Delivery of Somatostatin Peptide to Chronic Pancreatitis. Pharmaceutics 2022, 14, 2341. [Google Scholar] [CrossRef]
- Yan, I.K.; Shukla, N.; Borrelli, D.A.; Patel, T. Use of a Hollow Fiber Bioreactor to Collect Extracellular Vesicles from Cells in Culture. Methods Mol. Biol. 2018, 1740, 35–41. [Google Scholar]
- Desai, N.; Rana, D.; Salave, S.; Gupta, R.; Patel, P.; Karunakaran, B.; Sharma, A.; Giri, J.; Benival, D.; Kommineni, N. Chitosan: A Potential Biopolymer in Drug Delivery and Biomedical Applications. Pharmaceutics 2023, 15, 1313. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Koppisetti, H.; Pande, S.; Shukla, H.; Sirsat, B.; Ditani, A.S.; Mallick, P.P.; Kathar, U.; Kalia, K.; Tekade, R.K. Nanomedicine in the Treatment of Diabetic Nephropathy. Future Med. Chem. 2021, 13, 663–686. [Google Scholar] [CrossRef]
- Salave, S.; Rana, D.; Sharma, A.; Bharathi, K.; Gupta, R.; Khode, S.; Benival, D.; Kommineni, N. Polysaccharide Based Implantable Drug Delivery: Development Strategies, Regulatory Requirements, and Future Perspectives. Polysaccharides 2022, 3, 625–654. [Google Scholar] [CrossRef]
- Rajani, C.; Borisa, P.; Bagul, S.; Shukla, K.; Tambe, V.; Desai, N.; Tekade, R.K. Developmental Toxicity of Nanomaterials Used in Drug Delivery: Understanding Molecular Biomechanics and Potential Remedial Measures. In Pharmacokinetics and Toxicokinetic Considerations; Academic Press: Cambridge, MA, USA, 2022; Volume 2, pp. 685–725. [Google Scholar]
- Rana, D.; Salave, S.; Rawat, G.; Benival, D. Recent Trends in Drug Delivery and Emerging Biomedical Applications of Gelatin for Ophthalmic Indications. Macromol. Res. 2022, 30, 687–702. [Google Scholar] [CrossRef]
- Liu, L.; Bai, X.; Martikainen, M.V.; Kårlund, A.; Roponen, M.; Xu, W.; Hu, G.; Tasciotti, E.; Lehto, V.P. Cell Membrane Coating Integrity Affects the Internalization Mechanism of Biomimetic Nanoparticles. Nat. Commun. 2021, 12, 5726. [Google Scholar] [CrossRef]
- Zou, D.; Wu, Z.; Yi, X.; Hui, Y.; Yangb, G.; Liu, Y.; Tengjisi; Wang, H.; Brooks, A.; Wang, H.; et al. Nanoparticle Elasticity Regulates the Formation of Cell Membrane-Coated Nanoparticles and Their Nano-Bio Interactions. Proc. Natl. Acad. Sci. USA 2023, 120, e2214757120. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Yang, Y.; Guo, L.; Chen, M.; Zhou, X.; Zhao, Y.; Nie, D.; Gan, Y.; Zhang, X. Cell Membrane-Camouflaged Nanocarriers with Biomimetic Deformability of Erythrocytes for Ultralong Circulation and Enhanced Cancer Therapy. ACS Nano 2022, 16, 6527–6540. [Google Scholar] [CrossRef]
- Sung, S.Y.; Su, Y.L.; Cheng, W.; Hu, P.F.; Chiang, C.S.; Chen, W.T.; Hu, S.H. Graphene Quantum Dots-Mediated Theranostic Penetrative Delivery of Drug and Photolytics in Deep Tumors by Targeted Biomimetic Nanosponges. Nano Lett. 2019, 19, 69–81. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Gao, F.; Wei, J.J.; Qian, C.G.; Sun, M.J. Biomimetic Hybrid Nanozymes with Self-Supplied H+ and Accelerated O2 Generation for Enhanced Starvation and Photodynamic Therapy against Hypoxic Tumors. Nano Lett. 2019, 19, 4334–4342. [Google Scholar] [CrossRef]
- Chai, Z.; Ran, D.; Lu, L.; Zhan, C.; Ruan, H.; Hu, X.; Xie, C.; Jiang, K.; Li, J.; Zhou, J.; et al. Ligand-Modified Cell Membrane Enables the Targeted Delivery of Drug Nanocrystals to Glioma. ACS Nano 2019, 13, 5591–5601. [Google Scholar] [CrossRef]
- Li, J.; Ai, Y.; Wang, L.; Bu, P.; Sharkey, C.C.; Wu, Q.; Wun, B.; Roy, S.; Shen, X.; King, M.R. Targeted Drug Delivery to Circulating Tumor Cells via Platelet Membrane-Functionalized Particles. Biomaterials 2016, 76, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Qian, C.; Sun, W.; Wang, J.; Chen, Z.; Bomba, H.N.; Xin, H.; Shen, Q.; Gu, Z. Engineered Nanoplatelets for Enhanced Treatment of Multiple Myeloma and Thrombus. Adv. Mater. 2016, 28, 9573–9580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Li, F.; Lu, G.H.; Nie, W.; Zhang, L.; Lv, Y.; Bao, W.; Gao, X.; Wei, W.; Pu, K.; et al. Engineering Magnetosomes for Ferroptosis/Immunomodulation Synergism in Cancer. ACS Nano 2019, 13, 5662–5673. [Google Scholar] [CrossRef]
- Sun, Z.; Deng, G.; Peng, X.; Xu, X.; Liu, L.; Peng, J.; Ma, Y.; Zhang, P.; Wen, A.; Wang, Y.; et al. Intelligent Photothermal Dendritic Cells Restart the Cancer Immunity Cycle through Enhanced Immunogenic Cell Death. Biomaterials 2021, 279, 121228. [Google Scholar] [CrossRef]
- Xiao, P.; Wang, J.; Zhao, Z.; Liu, X.; Sun, X.; Wang, D.; Li, Y. Engineering Nanoscale Artificial Antigen-Presenting Cells by Metabolic Dendritic Cell Labeling to Potentiate Cancer Immunotherapy. Nano Lett. 2021, 21, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, T.; Endo, R.; Sasaki, S.; Takahashi, N.; Sato, Y.; Hyodo, M.; Hayakawa, Y.; Harashima, H. STING Agonist Loaded Lipid Nanoparticles Overcome Anti-PD-1 Resistance in Melanoma Lung Metastasis via NK Cell Activation. J. Immunother. Cancer 2021, 9, e002852. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, H.; Xie, B.R.; Qiu, W.X.; Zeng, J.Y.; Li, C.X.; Wan, S.S.; Zhang, L.; Liu, W.L.; Zhang, X.Z. Cancer Cell Membrane Camouflaged Cascade Bioreactor for Cancer Targeted Starvation and Photodynamic Therapy. ACS Nano 2017, 11, 7006–7018. [Google Scholar] [CrossRef]
- Rao, L.; Zhao, S.K.; Wen, C.; Tian, R.; Lin, L.; Cai, B.; Sun, Y.; Kang, F.; Yang, Z.; He, L.; et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv. Mater. 2020, 32, 2004853. [Google Scholar] [CrossRef]
- Yang, R.; Xu, J.; Xu, L.; Sun, X.; Chen, Q.; Zhao, Y.; Peng, R.; Liu, Z. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. [Google Scholar] [CrossRef]
- Gao, C.; Lin, Z.; Jurado-Sánchez, B.; Lin, X.; Wu, Z.; He, Q. Stem Cell Membrane-Coated Nanogels for Highly Efficient In Vivo Tumor Targeted Drug Delivery. Small 2016, 12, 4056–4062. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, F.; Liu, T.; Shao, P.; Duan, L.; Yan, J.; Mu, X.; Jiang, J. Polydopamine Nanoparticles Camouflaged by Stem Cell Membranes for Synergistic Chemo-Photothermal Therapy of Malignant Bone Tumors. Int. J. Nanomed. 2020, 15, 10183–10197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, Y.; Bu, W.; Meng, L.; Wang, C.; Jin, N.; Chen, Y.; Ren, C.; Zhang, K.; Sun, H. Modification of Metal-Organic Framework Nanoparticles Using Dental Pulp Mesenchymal Stem Cell Membranes to Target Oral Squamous Cell Carcinoma. J. Colloid Interface Sci. 2021, 601, 650–660. [Google Scholar] [CrossRef]
- Jc Bose, R.; Uday Kumar, S.; Zeng, Y.; Afjei, R.; Robinson, E.; Lau, K.; Bermudez, A.; Habte, F.; Pitteri, S.J.; Sinclair, R.; et al. Tumor Cell-Derived Extracellular Vesicle-Coated Nanocarriers: An Efficient Theranostic Platform for the Cancer-Specific Delivery of Anti-MiR-21 and Imaging Agents. ACS Nano 2018, 12, 10817–10832. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, Y.; Ding, F.; Yang, J.; Li, J.; Gao, X.; Zhang, C.; Feng, J. Engineering Macrophage-Derived Exosomes for Targeted Chemotherapy of Triple-Negative Breast Cancer. Nanoscale 2020, 12, 10854–10862. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hu, W.; Chen, H.; Shou, X.; Ye, T.; Xu, Y. Cocktail Strategy Based on NK Cell-Derived Exosomes and Their Biomimetic Nanoparticles for Dual Tumor Therapy. Cancers 2019, 11, 1560. [Google Scholar] [CrossRef] [Green Version]
- Malyutin, A.G.; Easterday, R.; Lozovyy, Y.; Spilotros, A.; Cheng, H.; Sanchez-Felix, O.R.; Stein, B.D.; Morgan, D.G.; Svergun, D.I.; Dragnea, B.; et al. Viruslike Nanoparticles with Maghemite Cores Allow for Enhanced Mri Contrast Agents. Chem. Mater. 2015, 27, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Wang, Q.; Li, J.; Kwong, C.H.T.; Wei, J.; Xie, B.; Lu, S.; Lee, S.M.Y.; Wang, R. In Vivo Hitchhiking of Immune Cells by Intracellular Self-Assembly of Bacteria-Mimetic Nanomedicine for Targeted Therapy of Melanoma. Sci. Adv. 2022, 8, eabn1805. [Google Scholar] [CrossRef]
- Zhou, J.; Karshalev, E.; Mundaca-Uribe, R.; de Esteban-Fernández Ávila, B.; Krishnan, N.; Xiao, C.; Ventura, C.J.; Gong, H.; Zhang, Q.; Gao, W.; et al. Physical Disruption of Solid Tumors by Immunostimulatory Microrobots Enhances Antitumor Immunity. Adv. Mater. 2021, 33, 2103505. [Google Scholar] [CrossRef]
- Harris, J.C.; Scully, M.A.; Day, E.S. Cancer Cell Membrane-Coated Nanoparticles for Cancer Management. Cancers 2019, 11, 1836. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Salave, S.; Rana, D.; Karunakaran, B.; Butreddy, A.; Benival, D.; Kommineni, N.; Mousavifar, L.; Gupta, R.; Salave, S.; et al. Versatility of Liposomes for Antisense Oligonucleotide Delivery: A Special Focus on Various Therapeutic Areas. Pharmaceutics 2023, 15, 1435. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Gao, W.; Zhang, L. Cell Membrane Coating Nanotechnology. Adv. Mater. 2018, 30, e1706759. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Cabe, M.H.; Ogle, S.D.; Sanchez, V.; Langert, K.A. Optimization of Critical Parameters for Coating of Polymeric Nanoparticles with Plasma Membrane Vesicles by Sonication. Sci. Rep. 2021, 11, 23996. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Hu, Y.; Luo, S.; Wang, Y.; Gong, T.; Sun, X.; Fu, Y.; Zhang, Z. Neutrophil-Mimicking Therapeutic Nanoparticles for Targeted Chemotherapy of Pancreatic Carcinoma. Acta Pharm. Sin. B 2019, 9, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Krishnamachary, B.; Barnett, J.D.; Chatterjee, S.; Chang, D.; Mironchik, Y.; Wildes, F.; Jaffee, E.M.; Nimmagadda, S.; Bhujwalla, Z.M. Human Cancer Cell Membrane Coated Biomimetic Nanoparticles Reduce Fibroblast-Mediated Invasion and Metastasis, and Induce T Cells. ACS Appl. Mater. Interfaces 2019, 11, 7850–7861. [Google Scholar] [CrossRef] [PubMed]
- Xuan, M.; Shao, J.; Dai, L.; He, Q.; Li, J. Macrophage Cell Membrane Camouflaged Mesoporous Silica Nanocapsules for In Vivo Cancer Therapy. Adv. Healthc. Mater. 2015, 4, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Jack Hu, C.M.; Fang, R.H.; Dehaini, D.; Carpenter, C.; Gao, W.; Zhang, L. Interfacial Interactions between Natural RBC Membranes and Synthetic Polymeric Nanoparticles. Nanoscale 2014, 6, 2730–2737. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.; Zhu, Q.; Wei, D.; Feng, J.; Yao, J.; Jiang, T.; Song, Q.; Wei, X.; Chen, H.; Gao, X.; et al. Nanoparticles Coated with Neutrophil Membranes Can Effectively Treat Cancer Metastasis. ACS Nano 2017, 11, 1397–1411. [Google Scholar] [CrossRef]
- Luk, B.T.; Jiang, Y.; Copp, J.A.; Hu, C.M.J.; Krishnan, N.; Gao, W.; Li, S.; Fang, R.H.; Zhang, L. Biomimetic Targeting of Nanoparticles to Immune Cell Subsets via Cognate Antigen Interactions. Mol. Pharm. 2018, 15, 3723–3728. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, W.; Li, Y.; Chang, J.; Tian, F.; Zhao, F.; Ma, Y.; Sun, J. Microfluidic Sonication to Assemble Exosome Membrane-Coated Nanoparticles for Immune Evasion-Mediated Targeting. Nano Lett. 2019, 19, 7836–7844. [Google Scholar] [CrossRef]
- Rao, L.; Cai, B.; Bu, L.L.; Liao, Q.Q.; Guo, S.S.; Zhao, X.Z.; Dong, W.F.; Liu, W. Microfluidic Electroporation-Facilitated Synthesis of Erythrocyte Membrane-Coated Magnetic Nanoparticles for Enhanced Imaging-Guided Cancer Therapy. ACS Nano 2017, 11, 3496–3505. [Google Scholar] [CrossRef]
- Andriola Silva, A.K.; Di Corato, R.; Pellegrino, T.; Chat, S.; Pugliese, G.; Luciani, N.; Gazeau, F.; Wilhelm, C. Cell-Derived Vesicles as a Bioplatform for the Encapsulation of Theranostic Nanomaterials. Nanoscale 2013, 5, 11374–11384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, J.; Chen, X.; Liu, W.; Chen, T. Cell Membrane Coating Technology: A Promising Strategy for Biomedical Applications. Nano-Micro Lett. 2019, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.; Park, W.; Park, S.B.; Rhim, W.K.; Han, D.K. Recent Trends in Cell Membrane-Cloaked Nanoparticles for Therapeutic Applications. Methods 2020, 177, 2–14. [Google Scholar] [CrossRef]
- Liu, L.; Yu, W.; Seitsonen, J.; Xu, W.; Lehto, V.P. Correct Identification of the Core-Shell Structure of Cell Membrane-Coated Polymeric Nanoparticles. Chemistry 2022, 28, e202200947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ye, Z.; Wei, L.; Luo, H.; Xiao, L. Cell Membrane-Coated Porphyrin Metal-Organic Frameworks for Cancer Cell Targeting and O2-Evolving Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 39594–39602. [Google Scholar] [CrossRef]
- Johnson, D.T.; Zhou, J.; Kroll, A.V.; Fang, R.H.; Yan, M.; Xiao, C.; Chen, X.; Kline, J.; Zhang, L.; Zhang, D.E. Acute Myeloid Leukemia Cell Membrane-Coated Nanoparticles for Cancer Vaccination Immunotherapy. Leukemia 2021, 36, 994–1005. [Google Scholar] [CrossRef]
- Wang, D.; Dong, H.; Li, M.; Cao, Y.; Yang, F.; Zhang, K.; Dai, W.; Wang, C.; Zhang, X. Erythrocyte-Cancer Hybrid Membrane Camouflaged Hollow Copper Sulfide Nanoparticles for Prolonged Circulation Life and Homotypic-Targeting Photothermal/Chemotherapy of Melanoma. ACS Nano 2018, 12, 5241–5252. [Google Scholar] [CrossRef]
- Ren, X.; Zheng, R.; Fang, X.; Wang, X.; Zhang, X.; Yang, W.; Sha, X. Red Blood Cell Membrane Camouflaged Magnetic Nanoclusters for Imaging-Guided Photothermal Therapy. Biomaterials 2016, 92, 13–24. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Liu, X.; Tian, Y.; Guo, H.; Jiang, T.; Luo, Z.; Jin, K.; Kuai, X.; Liu, Y.; et al. Enhanced Photothermal Therapy of Biomimetic Polypyrrole Nanoparticles through Improving Blood Flow Perfusion. Biomaterials 2017, 143, 130–141. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.M.J.; Fang, R.H.; Luk, B.T.; Su, J.; Zhang, L. Surface Functionalization of Gold Nanoparticles with Red Blood Cell Membranes. Adv. Mater. 2013, 25, 3549–3553. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.L.; Zou, M.Z.; Qin, S.Y.; Cheng, Y.J.; Ma, Y.H.; Sun, Y.X.; Zhang, X.Z. Recent Advances of Cell Membrane-Coated Nanomaterials for Biomedical Applications. Adv. Funct. Mater. 2020, 30, 202003559. [Google Scholar] [CrossRef]
- Nie, D.; Dai, Z.; Li, J.; Yang, Y.; Xi, Z.; Wang, J.; Zhang, W.; Qian, K.; Guo, S.; Zhu, C.; et al. Cancer-Cell-Membrane-Coated Nanoparticles with a Yolk-Shell Structure Augment Cancer Chemotherapy. Nano Lett. 2020, 20, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Bidkar, A.P.; Sanpui, P.; Ghosh, S.S. Transferrin-Conjugated Red Blood Cell Membrane-Coated Poly(Lactic-Co-Glycolic Acid) Nanoparticles for the Delivery of Doxorubicin and Methylene Blue. ACS Appl. Nano Mater. 2020, 3, 3807–3819. [Google Scholar] [CrossRef]
- Rao, L.; Meng, Q.F.; Bu, L.L.; Cai, B.; Huang, Q.; Sun, Z.J.; Zhang, W.F.; Li, A.; Guo, S.S.; Liu, W.; et al. Erythrocyte Membrane-Coated Upconversion Nanoparticles with Minimal Protein Adsorption for Enhanced Tumor Imaging. ACS Appl. Mater. Interfaces 2017, 9, 2159–2168. [Google Scholar] [CrossRef]
- Vuckovic, D.; Dagley, L.F.; Purcell, A.W.; Emili, A. Membrane Proteomics by High Performance Liquid Chromatography-Tandem Mass Spectrometry: Analytical Approaches and Challenges. Proteomics 2013, 13, 404–423. [Google Scholar] [CrossRef]
- Li, Z.; Cai, H.; Li, Z.; Ren, L.; Ma, X.; Zhu, H.; Gong, Q.; Zhang, H.; Gu, Z.; Luo, K. A Tumor Cell Membrane-Coated Self-Amplified Nanosystem as a Nanovaccine to Boost the Therapeutic Effect of Anti-PD-L1 Antibody. Bioact. Mater. 2022, 21, 299–312. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Zheng, D.W.; Zhang, M.K.; Yu, W.Y.; Qiu, W.X.; Hu, J.J.; Feng, J.; Zhang, X.Z. Preferential Cancer Cell Self-Recognition and Tumor Self-Targeting by Coating Nanoparticles with Homotypic Cancer Cell Membranes. Nano Lett. 2016, 16, 5895–5901. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Li, Z.; Li, D.; He, F.; Wang, K.; Tian, J.; Zhao, X. Biomimetic Camouflaged Nanoparticle-Based Folfirinox Platform for Optimizing Clinical Pancreatic Cancer Treatment. Nano Today 2023, 48, 101733. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, X.; Li, Q.; Shen, Y.; Zhou, T.; Liu, X. Macrophage-Evading and Tumor-Specific Apoptosis Inducing Nanoparticles for Targeted Cancer Therapy. Acta Pharm. Sin. B 2023, 13, 327–343. [Google Scholar] [CrossRef]
- Liu, L.; Pan, D.; Chen, S.; Martikainen, M.V.; Kårlund, A.; Ke, J.; Pulkkinen, H.; Ruhanen, H.; Roponen, M.; Käkelä, R.; et al. Systematic Design of Cell Membrane Coating to Improve Tumor Targeting of Nanoparticles. Nat. Commun. 2022, 13, 6181. [Google Scholar] [CrossRef]
- Salave, S.; Rana, D.; Benival, D. Peptide Functionalised Nanocarriers for Bone Specific Delivery of PTH (1-34) in Osteoporosis. Curr. Nanomed. 2021, 11, 142–148. [Google Scholar] [CrossRef]
- Salave, S.; Rana, D.; Kumar, H.; Kommineni, N.; Benival, D. Anabolic Peptide-Enriched Stealth Nanoliposomes for Effective Anti-Osteoporotic Therapy. Pharmaceutics 2022, 14, 2417. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Y.; Fan, Z.; Cheng, H. Cell Membrane Bioconjugation and Membrane-Derived Nanomaterials for Immunotherapy. Bioconjug. Chem. 2018, 29, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Pashuck, E.T.; Stevens, M.M. Achieving Controlled Biomolecule-Biomaterial Conjugation. Chem. Rev. 2018, 118, 7702–7743. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Wang, S.; Duan, Y.; Zhang, Q.; Chen, M.S.; Gao, W.; Zhang, L. Emerging Approaches to Functionalizing Cell Membrane-Coated Nanoparticles. Biochemistry 2021, 60, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cheng, S.; Jin, Y.; Zhang, N.; Wang, Y. Membrane Engineering of Cell Membrane Biomimetic Nanoparticles for Nanoscale Therapeutics. Clin. Transl. Med. 2021, 11, e292. [Google Scholar] [CrossRef] [PubMed]
- Ak, G.; Hamarat Şanlıer, Ş. Erythrocyte Membrane Vesicles Coated Biomimetic and Targeted Doxorubicin Nanocarrier: Development, Characterization and in Vitro Studies. J. Mol. Struct. 2020, 1205, 127664. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, D.; Song, Q.; Wu, T.; Zhuang, X.; Bao, Y.; Kong, M.; Qi, Y.; Tan, S.; Zhang, Z. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano 2015, 9, 6918–6933. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, X.; Ruan, M.; Liu, W.; Song, R.; Dai, J.; Xue, W. Worm-Like Biomimetic Nanoerythrocyte Carrying SiRNA for Melanoma Gene Therapy. Small 2018, 14, 201803002. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.M.J.; Chen, K.N.H.; Luk, B.T.; Carpenter, C.W.; Gao, W.; Li, S.; Zhang, D.E.; Lu, W.; Zhang, L. Lipid-Insertion Enables Targeting Functionalization of Erythrocyte Membrane-Cloaked Nanoparticles. Nanoscale 2013, 5, 8884–8888. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Qian, H.; Huang, J.; Sha, H.; Zhang, H.; Yu, L.; Liu, B.; Hua, D.; Qian, X. Anti-EGFR-IRGD Recombinant Protein Modified Biomimetic Nanoparticles Loaded with Gambogic Acid to Enhance Targeting and Antitumor Ability in Colorectal Cancer Treatment. Int. J. Nanomed. 2018, 13, 4961–4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.M.; Xie, W.; Xiao, Y.S.; Suo, M.; Zan, M.H.; Liao, Q.Q.; Hu, X.J.; Chen, L.B.; Chen, B.; Wu, W.T.; et al. Erythrocyte Membrane-Coated Gold Nanocages for Targeted Photothermal and Chemical Cancer Therapy. Nanotechnology 2018, 29, 084002. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody Fragments as Nanoparticle Targeting Ligands: A Step in the Right Direction. Chem. Sci. 2016, 8, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Sun, H.; Meng, Q.; Yin, Q.; Zhang, P.; Zhang, Z.; Yu, H.; Li, Y. Bioinspired Nanoparticles with NIR-Controlled Drug Release for Synergetic Chemophotothermal Therapy of Metastatic Breast Cancer. Adv. Funct. Mater. 2016, 26, 7495–7506. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, X.; Zhang, Y.; Xu, J.; Xu, J.; Li, Y.; Min, H.; Shi, J.; Zhao, Y.; Wei, J.; et al. Engineering Biomimetic Platesomes for PH-Responsive Drug Delivery and Enhanced Antitumor Activity. Adv. Mater. 2019, 31, 201900795. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, A.; Jiang, L.; Gu, Y.; Liu, J. Hybrid Membrane-Coated Biomimetic Nanoparticles (HM@BNPs): A Multifunctional Nanomaterial for Biomedical Applications. Biomacromolecules 2021, 22, 3149–3167. [Google Scholar] [CrossRef]
- Chen, H.Y.; Deng, J.; Wang, Y.; Wu, C.Q.; Li, X.; Dai, H.W. Hybrid Cell Membrane-Coated Nanoparticles: A Multifunctional Biomimetic Platform for Cancer Diagnosis and Therapy. Acta Biomater. 2020, 112, 1–13. [Google Scholar] [CrossRef]
- Liu, W.L.; Zou, M.Z.; Liu, T.; Zeng, J.Y.; Li, X.; Yu, W.Y.; Li, C.X.; Ye, J.J.; Song, W.; Feng, J.; et al. Cytomembrane Nanovaccines Show Therapeutic Effects by Mimicking Tumor Cells and Antigen Presenting Cells. Nat. Commun. 2019, 10, 3199. [Google Scholar] [CrossRef] [Green Version]
- Dehaini, D.; Wei, X.; Fang, R.H.; Masson, S.; Angsantikul, P.; Luk, B.T.; Zhang, Y.; Ying, M.; Jiang, Y.; Kroll, A.V.; et al. Erythrocyte-Platelet Hybrid Membrane Coating for Enhanced Nanoparticle Functionalization. Adv. Mater. 2017, 29, 201606209. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.L.; Zou, M.Z.; Liu, T.; Zeng, J.Y.; Li, X.; Yu, W.Y.; Li, C.X.; Ye, J.J.; Song, W.; Feng, J.; et al. Expandable Immunotherapeutic Nanoplatforms Engineered from Cytomembranes of Hybrid Cells Derived from Cancer and Dendritic Cells. Adv. Mater. 2019, 31, 201900499. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, G.; Wu, W.; Wang, J.; Hu, J.; Mao, J.; Chu, P.K.; Bai, H.; Tang, G. A Hybrid Eukaryotic-Prokaryotic Nanoplatform with Photothermal Modality for Enhanced Antitumor Vaccination. Adv. Mater. 2020, 32, 201908185. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Pan, H.; Li, W.; Chen, Z.; Ma, A.; Yin, T.; Liang, R.; Chen, F.; Ma, Y.; Jin, Y.; et al. T Cell Membrane Mimicking Nanoparticles with Bioorthogonal Targeting and Immune Recognition for Enhanced Photothermal Therapy. Adv. Sci. 2019, 6, 201900251. [Google Scholar] [CrossRef]
- Zhang, Q.; Wei, W.; Wang, P.; Zuo, L.; Li, F.; Xu, J.; Xi, X.; Gao, X.; Ma, G.; Xie, H.Y. Biomimetic Magnetosomes as Versatile Artificial Antigen-Presenting Cells to Potentiate T-Cell-Based Anticancer Therapy. ACS Nano 2017, 11, 10724–10732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, L.J.; Wang, S.M.; Yang, J.; Lu, G.H.; Luo, N.N.; Gao, X.Y.; Ma, G.H.; Xie, H.Y.; Wei, W. Construction of a Biomimetic Magnetosome and Its Application as a SiRNA Carrier for High-Performance Anticancer Therapy. Adv. Funct. Mater. 2018, 28, 1703326. [Google Scholar] [CrossRef]
- Price, N.L.; Goyette-Desjardins, G.; Nothaft, H.; Valguarnera, E.; Szymanski, C.M.; Segura, M.; Feldman, M.F. Glycoengineered Outer Membrane Vesicles: A Novel Platform for Bacterial Vaccines. Sci. Rep. 2016, 6, 24931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, P.; Liu, X.; Chen, X.; Liu, C.; Zhang, Y.; Chu, C.; Wang, J.; Wang, X.; Chen, X.; Liu, G. Genetically Engineered Cell Membrane Nanovesicles for Oncolytic Adenovirus Delivery: A Versatile Platform for Cancer Virotherapy. Nano Lett. 2019, 19, 2993–3001. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Muthukumaran, P.; Jayakumar, M.K.G.; Lisse, D.; Masurkar, N.D.; Xu, C.; Chan, J.M.; Drum, C.L. Surface Protein Engineering Increases the Circulation Time of a Cell Membrane-Based Nanotherapeutic. Nanomedicine 2019, 18, 169–178. [Google Scholar] [CrossRef]
- Pereira-Veiga, T.; Schneegans, S.; Pantel, K.; Wikman, H. Circulating Tumor Cell-Blood Cell Crosstalk: Biology and Clinical Relevance. Cell Rep. 2022, 40, 111298. [Google Scholar] [CrossRef]
- Huang, X.; Hu, X.; Song, S.; Mao, D.; Lee, J.; Koh, K.; Zhu, Z.; Chen, H. Triple-Enhanced Surface Plasmon Resonance Spectroscopy Based on Cell Membrane and Folic Acid Functionalized Gold Nanoparticles for Dual-Selective Circulating Tumor Cell Sensing. Sens. Actuators B Chem. 2020, 305, 127543. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Cui, T.; Ma, M.; Ren, J.; Qu, X. A Nature-Inspired Metal-Organic Framework Discriminator for Differential Diagnosis of Cancer Cell Subtypes. Angew. Chem. Int. Ed. Engl. 2021, 60, 15436–15444. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Liu, X.; Sang, Y.; Zhang, L.; Ren, J.; Qu, X. Cell Membrane-Camouflaged Liposomes for Tumor Cell-Selective Glycans Engineering and Imaging in Vivo. Proc. Natl. Acad. Sci. USA 2021, 118, 2022769118. [Google Scholar] [CrossRef]
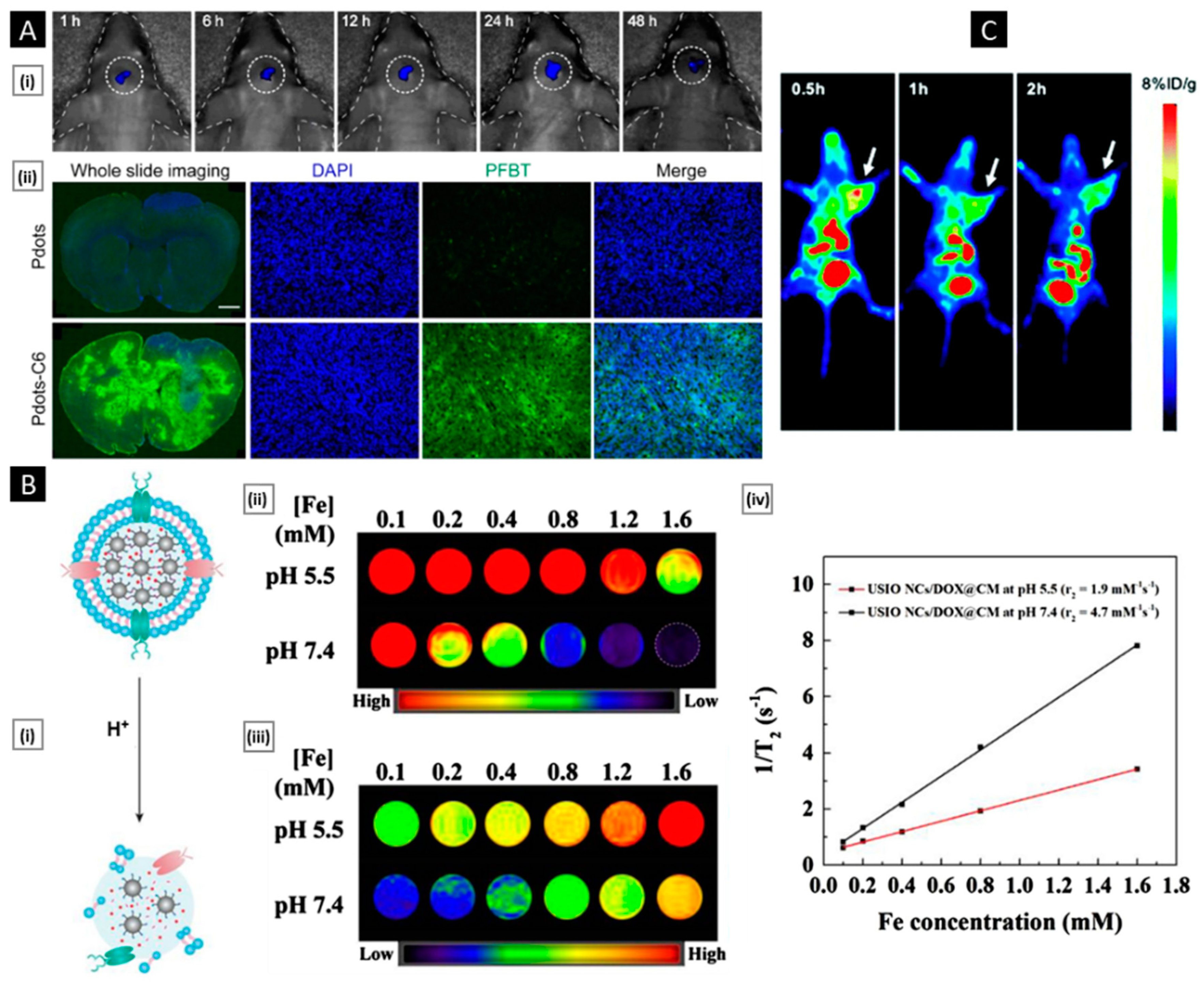
- Men, X.; Geng, X.; Zhang, Z.; Chen, H.; Du, M.; Chen, Z.; Liu, G.; Wu, C.; Yuan, Z. Biomimetic Semiconducting Polymer Dots for Highly Specific NIR-II Fluorescence Imaging of Glioma. Mater. Today Bio. 2022, 16, 100383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, M.; Chi, S.; Zhu, M.; Wang, C.; Liu, Z. Brain Tumor Cell Membrane-Coated Lanthanide-Doped Nanoparticles for NIR-IIb Luminescence Imaging and Surgical Navigation of Glioma. Adv. Healthc. Mater. 2022, 11, 202200521. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, H.; Chen, Z.; Lan, M. Ultrasmall Superparamagnetic Iron Oxide Nanoparticles for Magnetic Resonance Imaging Contrast Agent. J. Nanosci. Nanotechnol. 2014, 14, 210–220. [Google Scholar] [CrossRef]
- Jia, L.; Li, X.; Liu, H.; Xia, J.; Shi, X.; Shen, M. Ultrasound-Enhanced Precision Tumor Theranostics Using Cell Membrane-Coated and PH-Responsive Nanoclusters Assembled from Ultrasmall Iron Oxide Nanoparticles. Nano Today 2021, 36, 101022. [Google Scholar] [CrossRef]
- Du, K.; Feng, J.; Gao, X.; Zhang, H. Nanocomposites Based on Lanthanide-Doped Upconversion Nanoparticles: Diverse Designs and Applications. Light Sci. Appl. 2022, 11, 222. [Google Scholar] [CrossRef]
- Fang, H.; Li, M.; Liu, Q.; Gai, Y.; Yuan, L.; Wang, S.; Zhang, X.; Ye, M.; Zhang, Y.; Gao, M.; et al. Ultra-Sensitive Nanoprobe Modified with Tumor Cell Membrane for UCL/MRI/PET Multimodality Precise Imaging of Triple-Negative Breast Cancer. Nano-Micro Lett. 2020, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Fang, H.; Liu, Q.; Gai, Y.; Yuan, L.; Wang, S.; Li, H.; Hou, Y.; Gao, M.; Lan, X. Red Blood Cell Membrane-Coated Upconversion Nanoparticles for Pretargeted Multimodality Imaging of Triple-Negative Breast Cancer. Biomater. Sci. 2020, 8, 1802–1814. [Google Scholar] [CrossRef]
- Ferrel, C.; Rayamajhi, S.; Nguyen, T.; Marasini, R.; Saravanan, T.; Deba, F.; Aryal, S. Re-Engineering a Liposome with Membranes of Red Blood Cells for Drug Delivery and Diagnostic Applications. ACS Appl. Bio Mater. 2021, 4, 6974–6981. [Google Scholar] [CrossRef]
- Qu, Y.; Chu, B.; Wei, X.; Chen, Y.; Yang, Y.; Hu, D.; Huang, J.; Wang, F.; Chen, M.; Zheng, Y.; et al. Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Multiple Myeloma Based on Bone Marrow Homing. Adv. Mater. 2022, 34, 202107883. [Google Scholar] [CrossRef]
- Gao, G.; Sun, X.; Liang, G. Nanoagent-Promoted Mild-Temperature Photothermal Therapy for Cancer Treatment. Adv. Funct. Mater. 2021, 31, 2100738. [Google Scholar] [CrossRef]
- Yuan, A.; Ruan, L.; Jia, R.; Wang, X.; Wu, L.; Cao, J.; Qi, X.; Wei, Y.; Shen, S. Tumor Exosome-Mimicking Iron Oxide Nanoparticles for Near Infrared-Responsive Drug Delivery. ACS Appl. Nano Mater. 2022, 5, 996–1002. [Google Scholar] [CrossRef]
- Zhang, C.; Song, J.; Lou, L.; Qi, X.; Zhao, L.; Fan, B.; Sun, G.; Lv, Z.; Fan, Z.; Jiao, B.; et al. Doxorubicin-loaded Nanoparticle Coated with Endothelial Cells-derived Exosomes for Immunogenic Chemotherapy of Glioblastoma. Bioeng. Transl. Med. 2021, 6, 10203. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Lei, Q.; Zhang, X.Z. Recent Advances in Photonanomedicines for Enhanced Cancer Photodynamic Therapy. Prog. Mater. Sci. 2020, 114, 100685. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, D.; Cao, Y.; Wang, Y.; Wang, F.; Zhang, F.; Zheng, S. Biodegradable Hypocrellin B Nanoparticles Coated with Neutrophil Membranes for Hepatocellular Carcinoma Photodynamics Therapy Effectively via JUNB/ROS Signaling. Int. Immunopharmacol. 2021, 99, 107624. [Google Scholar] [CrossRef]
- Pan, W.L.; Tan, Y.; Meng, W.; Huang, N.H.; Zhao, Y.B.; Yu, Z.Q.; Huang, Z.; Zhang, W.H.; Sun, B.; Chen, J.X. Microenvironment-Driven Sequential Ferroptosis, Photodynamic Therapy, and Chemotherapy for Targeted Breast Cancer Therapy by a Cancer-Cell-Membrane-Coated Nanoscale Metal-Organic Framework. Biomaterials 2022, 283, 121449. [Google Scholar] [CrossRef]
- Desai, N.; Hasan, U.; Jeyashree, K.; Mani, R.; Chauhan, M.; Basu, S.M.; Giri, J. Biomaterial-Based Platforms for Modulating Immune Components against Cancer and Cancer Stem Cells. Acta Biomater. 2023, 161, 1–36. [Google Scholar] [CrossRef]
- Ma, X.; Kuang, L.; Yin, Y.; Tang, L.; Zhang, Y.; Fan, Q.; Wang, B.; Dong, Z.; Wang, W.; Yin, T.; et al. Tumor-Antigen Activated Dendritic Cell Membrane-Coated Biomimetic Nanoparticles with Orchestrating Immune Responses Promote Therapeutic Efficacy against Glioma. ACS Nano 2023, 17, 2341–2355. [Google Scholar] [CrossRef]
- Ma, J.; Chen, C.C.; Li, M. Macrophages/Microglia in the Glioblastoma Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 5775. [Google Scholar] [CrossRef]
- Gao, X.; Li, S.; Ding, F.; Liu, X.; Wu, Y.; Li, J.; Feng, J.; Zhu, X.; Zhang, C. A Virus-Mimicking Nucleic Acid Nanogel Reprograms Microglia and Macrophages for Glioblastoma Therapy. Adv. Mater. 2021, 33, 2006116. [Google Scholar] [CrossRef]
- Huang, X.; Lu, Y.; Guo, M.; Du, S.; Han, N. Recent Strategies for Nano-Based PTT Combined with Immunotherapy: From a Biomaterial Point of View. Theranostics 2021, 11, 7546–7569. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Bian, J.; Wei, J.; Wang, Z.; Zhou, Z.; Li, Z.; Sun, M. Converting Primary Tumor towards an in Situ STING-Activating Vaccine via a Biomimetic Nanoplatform against Recurrent and Metastatic Tumors. Nano Today 2021, 38, 101109. [Google Scholar] [CrossRef]
- Xiong, J.; Wu, M.; Chen, J.; Liu, Y.; Chen, Y.; Fan, G.; Liu, Y.; Cheng, J.; Wang, Z.; Wang, S.; et al. Cancer-Erythrocyte Hybrid Membrane-Camouflaged Magnetic Nanoparticles with Enhanced Photothermal-Immunotherapy for Ovarian Cancer. ACS Nano 2021, 15, 19756–19770. [Google Scholar] [CrossRef] [PubMed]
- Aytar Çelik, P.; Erdogan-Gover, K.; Barut, D.; Enuh, B.M.; Amasya, G.; Sengel-Türk, C.T.; Derkus, B.; Çabuk, A. Bacterial Membrane Vesicles as Smart Drug Delivery and Carrier Systems: A New Nanosystems Tool for Current Anticancer and Antimicrobial Therapy. Pharmaceutics 2023, 15, 1052. [Google Scholar] [CrossRef] [PubMed]









| Core | Delivery Cargo | Surface Modification | Application | Ref. |
|---|---|---|---|---|
| Blood Cells | ||||
| PEG nanogel | Doxorubicin (DOX) | - | Passive tumor targeting | [101] |
| Mesoporous silica | Graphene quantum dots; docetaxel | Cetuximab | PTT; light-triggered drug release | [102] |
| Manganese oxide | Chlorin e6; glucose oxidase | - | Tumor starvation; PDT | [103] |
| Nanocrystals | Docetaxel | cRGD peptide | Tumor targeted delivery | [104] |
| Silica | - | TNF-related apoptosis-inducing ligand | CTC-targeted delivery | [105] |
| Acetylated dextran | Bortezomib | Tissue plasminogen activator; alendronate | Bone-targeted delivery | [106] |
| Liposome | Emtansine | - | Metastatic tumor targeting | [31] |
| Iron oxide | - | TGF-β inhibitor (SB505124); anti-PD-1 antibody | Magnetically guided immune stimulation and ferroptosis | [107] |
| DSPE-PEG | IR-797 | - | PPT; ICD-mediated immune stimulation | [108] |
| poly(lactic-co-glycolic acid) (PLGA) | Imiquimod | Anti-CD3 antibody | Local immune stimulation; T cell-directed tumor antigen presentation | [109] |
| PLGA | 4,4′,4″,4‴-(Porphine-5,10,15,20- tetrayl) tetrakis(benzoic acid) | - | PDT; M1 macrophage polarization | [35] |
| Lipid NPs | Cyclic di-GMP | - | Immunogenic reprogramming of TME | [110] |
| MOF (PCN-224) | Glucose oxidase; catalase | - | Homotypic tumor-targeted PDT; starvation therapy | [111] |
| Iron oxide | - | Signal regulatory protein α | Magnetically guided immune stimulation; M1 macrophage polarization | [112] |
| PLGA | Imiquimod | Mannose | Antigen-presenting cell-targeted tumor antigen delivery | [113] |
| Gelatin nanogel | DOX | - | Tumor targeted delivery | [114] |
| Polydopamine NPs | SN-38 | - | PTT; acidic TME-responsive drug delivery | [115] |
| MOF | DOX | - | Chemokine-mediated active targeting | [116] |
| Gold-iron oxide | Anti-miR-21 | ICG | Homotypic tumor-targeted delivery, MR imaging | [117] |
| PLGA | DOX | Mesenchymal-epithelial transition factor binding peptide | Prolonged circulation lifetime; tumor-targeted delivery | [118] |
| Tyrosine-coupled dendrimers | let-7a mimics | - | Tumor targeted delivery | [119] |
| Gold-coated maghemite | - | - | MR imaging | [120] |
| Quantum dots | Anticancer drugs (DOX, cisplatin, and 5-fluorouracil) and/or siRNA cocktail and/or anticancer toxins | SP94 targeting peptide; histidine-rich fusogenic peptide (for endosomal escape) | Tumor targeted delivery, multimodal anticancer effect | [66] |
| β-cyclodextrin-modified GNPs and adamantane-modified Gold NPs | - | - | Inflammation-targeted PTT; ant-tumor immunotherapy; TME modulation | [121] |
| Titanium dioxide-coated magnesium Janus micromotor | - | - | Local tumor disruption; immune stimulation | [122] |
| Polyplex (PC7A) | CpG oligodeoxynucleotides (CpG 1826) | Maleimide | Tumor antigen capture and cross-presentation; immune stimulation | [84,85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, N.; Rana, D.; Pande, S.; Salave, S.; Giri, J.; Benival, D.; Kommineni, N. “Bioinspired” Membrane-Coated Nanosystems in Cancer Theranostics: A Comprehensive Review. Pharmaceutics 2023, 15, 1677. https://doi.org/10.3390/pharmaceutics15061677
Desai N, Rana D, Pande S, Salave S, Giri J, Benival D, Kommineni N. “Bioinspired” Membrane-Coated Nanosystems in Cancer Theranostics: A Comprehensive Review. Pharmaceutics. 2023; 15(6):1677. https://doi.org/10.3390/pharmaceutics15061677
Chicago/Turabian StyleDesai, Nimeet, Dhwani Rana, Shreya Pande, Sagar Salave, Jyotsnendu Giri, Derajram Benival, and Nagavendra Kommineni. 2023. "“Bioinspired” Membrane-Coated Nanosystems in Cancer Theranostics: A Comprehensive Review" Pharmaceutics 15, no. 6: 1677. https://doi.org/10.3390/pharmaceutics15061677