
Distribution and Activity of Sulfur-Metabolizing Bacteria along the Temperature Gradient in Phototrophic Mats of the Chilean Hot Spring Porcelana
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Site and Sample Collection
2.2. Determination of Inorganic Sulfur Species in Porcelana Thermal Water
2.3. DNA Extraction, PCR Amplification, Cloning, and Sequencing of the aprA and soxB Genes
2.4. Sulfur Metabolism Reconstruction from Publicly Available Porcelana Hot Spring Metagenomes and Metatranscriptomes
2.5. Taxonomic Description and Phylogenetic Reconstruction
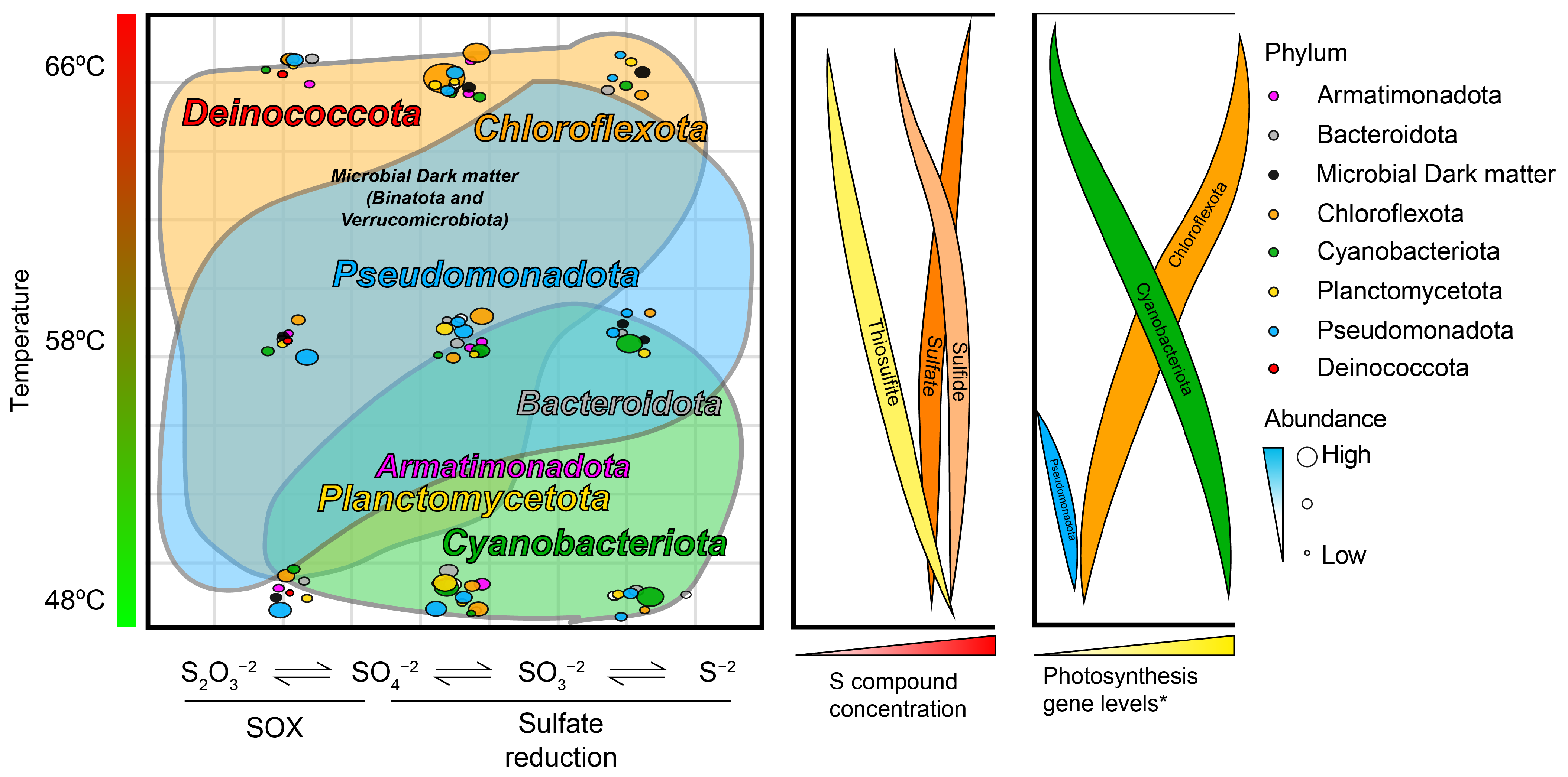
3. Results and Discussion
3.1. Changes in Sulfur Compounds along the Temperature Gradient at Porcelana Hot Spring
3.2. Key Active Sulfur-Metabolizing Organisms and Pathways in the Sulfur Cycle of Porcelana Revealed by Meta-Omics
3.3. Complementary Diversity Analysis of Sulfur-Metabolizing Organisms Using aprA and soxB Marker Genes
3.4. Phylogenetic Affiliation and Abundance of Sulfur-Metabolizing OTUs and MAGs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgan, J.W.; Anders, E. Chemical composition of Earth, Venus, and Mercury. Proc. Natl. Acad. Sci. USA 1980, 77, 6973–6977. [Google Scholar] [CrossRef] [PubMed]
- Fike, D.A.; Bradley, A.S.; Rose, C.V. Rethinking the Ancient Sulfur Cycle. Annu. Rev. Earth Planet. Sci. 2015, 43, 593–622. [Google Scholar] [CrossRef] [Green Version]
- Tassi, F.; Aguilera, F.; Darrah, T.; Vaselli, O.; Capaccioni, B.; Poreda, R.; Huertas, A.D. Fluid geochemistry of hydrothermal systems in the Arica-Parinacota, Tarapacá and Antofagasta regions (northern Chile). J. Volcanol. Geotherm. Res. 2010, 192, 1–15. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; McCleskey, R.B.; Ball, J.W. Sulfur geochemistry of hydrothermal waters in Yellowstone National Park: IV Acid-sulfate waters. Appl. Geochem. 2009, 24, 191–207. [Google Scholar] [CrossRef]
- Canfield, D.E.; Kristensen, E.; Thamdrup, B. The Sulfur Cycle. In Advances in Marine Biology; Canfield, D.E., Kristensen, E., Thamdrup, B., Eds.; Academic Press: Cambridge, MA, USA, 2005; Volume 48, pp. 313–381. ISBN 0065-2881. [Google Scholar]
- Bolhuis, H.; Cretoiu, M.S.; Stal, L.J. Molecular ecology of microbial mats. FEMS Microbiol. Ecol. 2014, 90, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Barajas, C.M.; Valencia-Cantero, E.; Santoyo, G. Microbial mat ecosystems: Structure types, functional diversity, and biotechnological application. Electron. J. Biotechnol. 2018, 31, 48–56. [Google Scholar] [CrossRef]
- Brazelton, W.J.; Schrenk, M.O.; Kelley, D.S.; Baross, J.A. Methane- and Sulfur-Metabolizing Microbial Communities Dominate the Lost City Hydrothermal Field Ecosystem. Appl. Environ. Microbiol. 2006, 72, 6257–6270. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; She, W.; Wu, G.; Yang, J.; Phurbu, D.; Jiang, H. Influence of Temperature and Sulfate Concentration on the Sulfate/Sulfite Reduction Prokaryotic Communities in the Tibetan Hot Springs. Microorganisms 2021, 9, 583. [Google Scholar] [CrossRef]
- Rojas-Gätjens, D.; Arce-Rodríguez, A.; Puente-Sánchez, F.; Avendaño, R.; Libby, E.; Mora-Amador, R.; Rojas-Jimenez, K.; Fuentes-Schweizer, P.; Pieper, D.H.; Chavarría, M. Temperature and elemental sulfur shape microbial communities in two extremely acidic aquatic volcanic environments. Extremophiles 2021, 25, 85–99. [Google Scholar] [CrossRef]
- Schmidt, A. Sulfur metabolism in cyanobacteria. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1988; Volume 167, pp. 572–583. ISBN 978-0-12-182068-8. [Google Scholar] [CrossRef]
- Kharwar, S.; Bhattacharjee, S.; Chakraborty, S.; Mishra, A.K. Regulation of sulfur metabolism, homeostasis and adaptive responses to sulfur limitation in cyanobacteria. Biologia 2021, 76, 2811–2835. [Google Scholar] [CrossRef]
- Blankenship, R.E. Electron transport in green photosynthetic bacteria. Photosynth. Res. 1985, 6, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Revsbech, N.P.; Ward, D.M.; Castenholz, R.W. Obligately phototrophic Chloroflexus: Primary production in anaerobic hot spring microbial mats. Arch. Microbiol. 1987, 147, 80–87. [Google Scholar] [CrossRef]
- Kanno, N.; Haruta, S.; Hanada, S. Sulfide-dependent Photoautotrophy in the Filamentous Anoxygenic Phototrophic Bacterium, Chloroflexus aggregans. Microbes Environ. 2019, 34, 304–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Liu, F.; Fang, W.; Yang, T.; Chen, G.-H.; He, Z.; Wang, S. Microbial sulfur metabolism and environmental implications. Sci. Total Environ. 2021, 778, 146085. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Hügler, M.; Ward, D.M.; Bryant, D.A. The Dark Side of the Mushroom Spring Microbial Mat: Life in the Shadow of Chlorophototrophs. II. Metabolic Functions of Abundant Community Members Predicted from Metagenomic Analyses. Front. Microbiol. 2017, 8, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, L.; Chi, A.; Beard, S.; Orell, A.; Guiliani, N.; Shabanowitz, J.; Hunt, D.F.; Jerez, C.A. Genomics, metagenomics and proteomics in biomining microorganisms. Biotechnol. Adv. 2006, 24, 197–211. [Google Scholar] [CrossRef]
- Yuan, H.; Yuan, J.; You, Y.; Zhang, B.; Wu, Y.; Huang, S.; Zhang, Y. Simultaneous ammonium and sulfate biotransformation driven by aeration: Nitrogen/sulfur metabolism and metagenome-based microbial ecology. Sci. Total Environ. 2021, 794, 148650. [Google Scholar] [CrossRef]
- Anitori, R.P.; Trott, C.; Saul, D.J.; Bergquist, P.L.; Walter, M.R.; Brugger, J.; Wülser, P.-A.; Foden, J.; Cavicchioli, R.; Bebout, B.M.; et al. A Culture-Independent Survey of the Bacterial Community in a Radon Hot Spring. Astrobiology 2002, 2, 255–270. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.E.; Cady, S.L.; Pace, N.R. Microbial Composition of Near-Boiling Silica-Depositing Thermal Springs throughout Yellowstone National Park. Appl. Environ. Microbiol. 2002, 68, 5123–5135. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Fukui, M. Molecular Characterization of Community Structures and Sulfur Metabolism within Microbial Streamers in Japanese Hot Springs. Appl. Environ. Microbiol. 2003, 69, 7044–7057. [Google Scholar] [CrossRef] [Green Version]
- Ferris, M.J.; Magnuson, T.S.; Fagg, J.A.; Thar, R.; Kühl, M.; Sheehan, K.B.; Henson, J.M. Microbially mediated sulphide production in a thermal, acidic algal mat community in Yellowstone National Park. Environ. Microbiol. 2003, 5, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, K.; Knittel, K.; Amann, R.; Fukui, M.; Matsuura, K. Sulfur-metabolizing bacterial populations in microbial mats of the Nakabusa hot spring, Japan. Syst. Appl. Microbiol. 2011, 34, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Klatt, C.G.; Inskeep, W.P.; Herrgard, M.J.; Jay, Z.J.; Rusch, D.B.; Tringe, S.G.; Parenteau, M.N.; Ward, D.M.; Boomer, S.M.; Bryant, D.A.; et al. Community Structure and Function of High-Temperature Chlorophototrophic Microbial Mats Inhabiting Diverse Geothermal Environments. Front. Microbiol. 2013, 4, 106. [Google Scholar] [CrossRef] [Green Version]
- Kappler, U.; Dahl, C. Enzymology and molecular biology of prokaryotic sulfite oxidation. FEMS Microbiol. Lett. 2001, 203, 1–9. [Google Scholar] [CrossRef]
- Meyer, B.; Kuever, J. Homology Modeling of Dissimilatory APS Reductases (AprBA) of Sulfur-Oxidizing and Sulfate-Reducing Prokaryotes. PLoS ONE 2008, 3, e1514. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.; Imhoff, J.F.; Kuever, J. Molecular analysis of the distribution and phylogeny of the soxB gene among sulfur-oxidizing bacteria—Evolution of the Sox sulfur oxidation enzyme system. Environ. Microbiol. 2007, 9, 2957–2977. [Google Scholar] [CrossRef] [Green Version]
- Green-Saxena, A.; Feyzullayev, A.; Hubert, C.R.J.; Kallmeyer, J.; Krueger, M.; Sauer, P.; Schulz, H.-M.; Orphan, V.J. Active sulfur cycling by diverse mesophilic and thermophilic microorganisms in terrestrial mud volcanoes of Azerbaijan. Environ. Microbiol. 2012, 14, 3271–3286. [Google Scholar] [CrossRef]
- Hügler, M.; Gärtner, A.; Imhoff, J.F. Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 2010, 73, 526–537. [Google Scholar] [CrossRef]
- Sakurai, H.; Ogawa, T.; Shiga, M.; Inoue, K. Inorganic sulfur oxidizing system in green sulfur bacteria. Photosynth. Res. 2010, 104, 163–176. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takai, K. Sulfur Metabolisms in Epsilon- and Gamma-Proteobacteria in Deep-Sea Hydrothermal Fields. Front. Microbiol. 2011, 2, 192. [Google Scholar] [CrossRef] [Green Version]
- Jaffer, Y.D.; Purushothaman, C.S.; Kumar, H.S.; Irfan, A.B.; Gireesh-Babu, P.; Ganie, P.A.; Bhat, R.A.H.; Vennila, A. A combined approach of 16S rRNA and a functional marker gene, soxB to reveal the diversity of sulphur-oxidising bacteria in thermal springs. Arch. Microbiol. 2019, 201, 951–967. [Google Scholar] [CrossRef]
- Petri, R.; Podgorsek, L.; Imhoff, J.F. Phylogeny and distribution of the soxB gene among thiosulfate-oxidizing bacteria. FEMS Microbiol. Lett. 2001, 197, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Fukui, M. Phylogenetic characterization of microbial mats and streamers from a Japanese alkaline hot spring with a thermal gradient. J. Gen. Appl. Microbiol. 2002, 48, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcamán-Arias, M.E.; Pedrós-Alió, C.; Tamames, J.; Fernández, C.; Pérez-Pantoja, D.; Vásquez, M.; Díez, B. Diurnal Changes in Active Carbon and Nitrogen Pathways Along the Temperature Gradient in Porcelana Hot Spring Microbial Mat. Front. Microbiol. 2018, 9, 2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, R.; Pedrós-Alió, C.; Díez, B. Bacterial composition of microbial mats in hot springs in Northern Patagonia: Variations with seasons and temperature. Extremophiles 2012, 17, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Alcamán, M.E.; Fernandez, C.; Delgado, A.; Bergman, B.; Díez, B. The cyanobacterium Mastigocladus fulfills the nitrogen demand of a terrestrial hot spring microbial mat. ISME J. 2015, 9, 2290–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, A. Standard Methods for the Examination of Water and Waste Water. Am. J. Public Health Nations Health 1966, 56, 387–388. [Google Scholar]
- Cline, J.D. Spectrophotometric determination of hydrogen sulfide in natural waters 1. Limnol. Oceanogr. 1969, 14, 454–458. [Google Scholar] [CrossRef]
- Lundquist, P.; Mårtensson, J.; Sörbo, B.; Ohman, S. Turbidimetry of inorganic sulfate, ester sulfate, and total sulfur in urine. Clin. Chem. 1980, 26, 1178–1181. [Google Scholar] [CrossRef]
- Koh, T.; Kitami, K.; Yonemura, Y. Spectrophotometric Determination of Thiosulfate by Its Oxidation with Iodate. Anal. Sci. 1991, 7, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.; Kuever, J. Molecular Analysis of the Diversity of Sulfate-Reducing and Sulfur-Oxidizing Prokaryotes in the Environment, Using aprA as Functional Marker Gene. Appl. Environ. Microbiol. 2007, 73, 7664–7679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcorta, J.; Alarcón-Schumacher, T.; Salgado, O.; Díez, B. Taxonomic Novelty and Distinctive Genomic Features of Hot Spring Cyanobacteria. Front. Genet. 2020, 11, 568223. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, btz848. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Prestat, E.; David, M.M.; Hultman, J.; Taş, N.; Lamendella, R.; Dvornik, J.; Mackelprang, R.; Myrold, D.D.; Jumpponen, A.; Tringe, S.G.; et al. FOAM (Functional Ontology Assignments for Metagenomes): A Hidden Markov Model (HMM) database with environmental focus. Nucleic Acids Res. 2014, 42, e145. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Hiraishi, A.; Kato, K.; Chiura, H.X.; Maki, Y.; Shimizu, A. Phylogenetic Evidence for the Existence of Novel Thermophilic Bacteria in Hot Spring Sulfur-Turf Microbial Mats in Japan. Appl. Environ. Microbiol. 1998, 64, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Schoonen, M.; Nordstrom, D.; Cunningham, K.; Ball, J. Sulfur geochemistry of hydrothermal waters in Yellowstone National Park: I. The origin of thiosulfate in hot spring waters. Geochim. Cosmochim. Acta 1998, 62, 3729–3743. [Google Scholar] [CrossRef]
- Podar, P.T.; Yang, Z.; Björnsdóttir, S.H.; Podar, M. Comparative Analysis of Microbial Diversity across Temperature Gradients in Hot Springs From Yellowstone and Iceland. Front. Microbiol. 2020, 11, 1625. [Google Scholar] [CrossRef] [PubMed]
- Macur, R.; Jay, Z.; Taylor, W.; Kozubal, M.; Kocar, B.; Inskeep, W. Microbial community structure and sulfur biogeochemistry in mildly-acidic sulfidic geothermal springs in Yellowstone National Park. Geobiology 2012, 11, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loy, A.; Duller, S.; Baranyi, C.; Mußmann, M.; Ott, J.; Sharon, I.; Béjà, O.; Le Paslier, D.; Dahl, C.; Wagner, M. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ. Microbiol. 2009, 11, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Pott, A.S.; Dahl, C. Sirohaem sulfite reductase and other proteins encoded by genes at the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 1998, 144, 1881–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Kubo, K.; Kamei, Y.; Kojima, H.; Fukui, M. Dissimilatory microbial sulfur and methane metabolism in the water column of a shallow meromictic lake. Syst. Appl. Microbiol. 2022, 45, 126320. [Google Scholar] [CrossRef]
- Murphy, C.L.; Sheremet, A.; Dunfield, P.F.; Spear, J.R.; Stepanauskas, R.; Woyke, T.; Elshahed, M.S.; Youssef, N.H. Genomic Analysis of the Yet-Uncultured Binatota Reveals Broad Methylotrophic, Alkane-Degradation, and Pigment Production Capacities. mBio 2021, 12, e00985-21. [Google Scholar] [CrossRef]
- Prieto-Barajas, C.M.; Alfaro-Cuevas, R.; Valencia-Cantero, E.; Santoyo, G. Effect of seasonality and physicochemical parameters on bacterial communities in two hot spring microbial mats from Araró, Mexico. Rev. Mex. Biodivers. 2017, 88, 616–624. [Google Scholar] [CrossRef]
- Watanabe, T.; Kojima, H.; Fukui, M. Identity of major sulfur-cycle prokaryotes in freshwater lake ecosystems revealed by a comprehensive phylogenetic study of the dissimilatory adenylylsulfate reductase. Sci. Rep. 2016, 6, 36262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X.; Zhen, Y.; Mi, T.; He, H.; Yu, Z. Microbial Diversity and Community Structure of Sulfate-Reducing and Sulfur-Oxidizing Bacteria in Sediment Cores from the East China Sea. Front. Microbiol. 2017, 8, 2133. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.S.; van Zyl, A.W.; van Hille, R.P.; Thomas, B.C.; Harrison, S.T.L.; Banfield, J.F. Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ. Microbiol. 2015, 17, 4929–4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, D.J.; Andreote, F.D.; Chaves, D.; Montaña, J.S.; Osorio-Forero, C.; Junca, H.; Zambrano, M.M.; Baena, S. Structural and Functional Insights from the Metagenome of an Acidic Hot Spring Microbial Planktonic Community in the Colombian Andes. PLoS ONE 2012, 7, e52069. [Google Scholar] [CrossRef] [Green Version]
- Bohorquez, L.C.; Delgado-Serrano, L.; López, G.; Osorio-Forero, C.; Klepac-Ceraj, V.; Kolter, R.; Junca, H.; Baena, S.; Zambrano, M.M. In-depth Characterization via Complementing Culture-Independent Approaches of the Microbial Community in an Acidic Hot Spring of the Colombian Andes. Microb. Ecol. 2012, 63, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berben, T.; Overmars, L.; Sorokin, D.Y.; Muyzer, G. Diversity and Distribution of Sulfur Oxidation-Related Genes in Thioalkalivibrio, a Genus of Chemolithoautotrophic and Haloalkaliphilic Sulfur-Oxidizing Bacteria. Front. Microbiol. 2019, 10, 160. [Google Scholar] [CrossRef]
- Vésteinsdóttir, H.; Reynisdóttir, D.B.; Örlygsson, J. Hydrogenophilus islandicus sp. nov., a thermophilic hydrogen-oxidizing bacterium isolated from an Icelandic hot spring. Int. J. Syst. Evol. Microbiol. 2011, 61, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, R.; Waberski, A.; Liesack, W.; Völker, H.; Wehmeyer, U.; Thomm, M. Hydrogenophilus hirschii sp. nov., a novel thermophilic hydrogen-oxidizing beta-proteobacterium isolated from Yellowstone National Park. Int. J. Syst. Evol. Microbiol. 2001, 51, 481–488. [Google Scholar] [CrossRef]
- Kimura, H.; Sugihara, M.; Yamamoto, H.; Patel, B.K.C.; Kato, K.; Hanada, S. Microbial community in a geothermal aquifer associated with the subsurface of the Great Artesian Basin, Australia. Extremophiles 2005, 9, 407–414. [Google Scholar] [CrossRef]
- Skirnisdottir, S.; Hreggvidsson, G.O.; Hjörleifsdottir, S.; Marteinsson, V.T.; Petursdottir, S.K.; Holst, O.; Kristjansson, J.K. Influence of Sulfide and Temperature on Species Composition and Community Structure of Hot Spring Microbial Mats. Appl. Environ. Microbiol. 2000, 66, 2835–2841. [Google Scholar] [CrossRef] [Green Version]
- Haouari, O.; Fardeau, M.-L.; Cayol, J.-L.; Casiot, C.; Elbaz-Poulichet, F.; Hamdi, M.; Joseph, M.; Ollivier, B. Desulfotomaculum hydrothermale sp. nov., a thermophilic sulfate-reducing bacterium isolated from a terrestrial Tunisian hot spring. Int. J. Syst. Evol. Microbiol. 2008, 58, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Karnauchow, T.M.; Jarrell, K.F.; Balkwill, D.L.; Drake, G.R.; Ringelberg, D.; Clarno, R.; Boone, D.R. Description of Two New Thermophilic Desulfotomaculum spp., Desulfotomaculum putei sp. nov., from a Deep Terrestrial Subsurface, and Desulfotomaculum luciae sp. nov., from a Hot Spring. Int. J. Syst. Evol. Microbiol. 1997, 47, 615–621. [Google Scholar] [CrossRef]
- Kopriva, S.; Büchert, T.; Fritz, G.; Suter, M.; Benda, R.; Schünemann, V.; Koprivova, A.; Schürmann, P.; Trautwein, A.X.; Kroneck, P.M.; et al. The Presence of an Iron-Sulfur Cluster in Adenosine 5′-Phosphosulfate Reductase Separates Organisms Utilizing Adenosine 5′-Phosphosulfate and Phosphoadenosine 5′-Phosphosulfate for Sulfate Assimilation. J. Biol. Chem. 2002, 277, 21786–21791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Tada, C.; Fukuda, Y.; Nakai, Y. Diversity of Sulfur-oxidizing Bacteria at the Surface of Cattle Manure Composting Assessed by an Analysis of the Sulfur Oxidation Gene soxB. Microbes Environ. 2020, 35, ME18066. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-L.; Yu, S.-L.; Gu, J.; Zhao, G.-F.; Chi, C.-Q. Filomicrobium insigne sp. nov., isolated from an oil-polluted saline soil. Int. J. Syst. Evol. Microbiol. 2009, 59, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kojima, H.; Fukui, M. Sulfuriferula multivorans gen. nov., sp. nov., isolated from a freshwater lake, reclassification of ‘Thiobacillus plumbophilus’ as Sulfuriferula plumbophilus sp. nov., and description of Sulfuricellaceae fam. nov. and Sulfuricellales ord. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 1504–1508. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.; Kuever, J. Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5′-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing prokaryotes. Microbiology 2007, 153, 3478–3498. [Google Scholar] [CrossRef] [Green Version]
- Hug, L.A.; Thomas, B.C.; Sharon, I.; Brown, C.T.; Sharma, R.; Hettich, R.L.; Wilkins, M.J.; Williams, K.H.; Singh, A.; Banfield, J.F. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. 2015, 18, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Inskeep, W.P.; Jay, Z.J.; Tringe, S.G.; Herrgård, M.J.; Rusch, D.B. The YNP metagenome project: Environmental parameters responsible for microbial distribution in the Yellowstone geothermal ecosystem. Front. Microbiol. 2013, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, C.B.; Stamps, B.W.; Lau, G.E.; Grasby, S.E.; Templeton, A.S.; Spear, J.R. Microbial Metabolic Redundancy Is a Key Mechanism in a Sulfur-Rich Glacial Ecosystem. Msystems 2020, 5, e00504-20. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Diversity Index | Sample 48_2013 | Sample 56.8_2011 | Sample 58_2013 | Sample 61.2_2011 | Sample 63.9_2011 | Sample 66_2013 | Sample 69.7_2011 |
|---|---|---|---|---|---|---|---|---|
| aprA | Simpson (1-D) | 0.112 | 0.056 | 0.718 | 0.409 | 0.383 | 0.675 | 0.413 |
| aprA | Shannon (H) | 0.291 | 0.149 | 1.391 | 0.855 | 0.802 | 1.213 | 0.762 |
| aprA | Evenness (J) | 0.181 | 0.135 | 0.863 | 0.531 | 0.498 | 0.753 | 0.549 |
| aprA | Richness | 5 | 3 | 5 | 5 | 5 | 5 | 4 |
| soxB | Simpson (1-D) | 0.499 | 0.368 | 0.32 | 0.147 | 0.336 | 0.313 | 0.436 |
| soxB | Shannon (H) | 0.692 | 0.555 | 0.504 | 0.278 | 0.519 | 0.492 | 0.628 |
| soxB | Evenness (J) | 0.992 | 0.801 | 0.721 | 0.402 | 0.749 | 0.71 | 0.906 |
| soxB | Richness | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Gene | OTU Clone | Relative Abundance | NCBI Nucleotide ID | GTDB R95 Taxonomy Best Hit 1 | % Aminoacid Identity | Matched Genome | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | - | Sample 48_2013 | Sample 56.8_2011 | Sample 58_2013 | Sample 61.2_2011 | Sample 63.9_2011 | Sample 66_2013 | Sample 69.7_2011 | ||||
| aprA | OTU-1 | 0.941 | 0.971 | 0.406 | 0.756 | 0.774 | 0.325 | 0.739 | KJ020275.1 | o_Burkholderiales f_Burkholderiaceae g_SCN-69-89 | 98.5 | GCA_001724855.1 |
| aprA | OTU-2 | 0.008 | 0.000 | 0.146 | 0.000 | 0.043 | 0.400 | 0.000 | KJ020276.1 | o_Burkholderiales f__SG8-39 g__SCGC-AG-212-J23 | 94.7 | GCA_004297625.1 |
| aprA | OTU-3 | 0.008 | 0.000 | 0.271 | 0.022 | 0.097 | 0.238 | 0.000 | KJ020277.1 | o_Burkholderiales f_Hydrogenophilaceae g_UBA6918 | 87.3 | GCA_002327405.1 |
| aprA | OTU-4 | 0.034 | 0.000 | 0.031 | 0.089 | 0.075 | 0.000 | 0.043 | KJ020278.1 | o_Thermodesulfovibrionales f_Thermodesulfovibrionaceae g_Thermodesulfovibrio | 97.9 | GCF_000020985.1 |
| aprA | OTU-5 | 0.008 | 0.019 | 0.146 | 0.100 | 0.011 | 0.025 | 0.196 | KJ020279.1 | f_Thermodesulfobacteriaceae g_Thermodesulfobacterium | 97.8 | GCF_000421605.1 |
| aprA | OTU-6 | 0.000 | 0.010 | 0.000 | 0.033 | 0.000 | 0.013 | 0.022 | KJ020280.1 | o_Desulfotomaculales f_Desulfovirgulaceae g_Desulfotomaculum_A | 93.2 | GCF_900129285.1 |
| soxB | OTU-1 | 0.511 | 0.764 | 0.800 | 0.914 | 0.784 | 0.811 | 0.678 | KJ026528.1 | o_Burkholderiales f_Rhodocyclaceae g_Tepidiphilus | 100.0 | GCF_001418245.1 * |
| soxB | OTU-2 | 0.489 | 0.236 | 0.200 | 0.086 | 0.216 | 0.189 | 0.322 | KJ026529.1 | o_Rhizobiales f_Hyphomicrobiaceae g_Filomicrobium | 80.4 | GCF_900104305.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konrad, R.; Vergara-Barros, P.; Alcorta, J.; Alcamán-Arias, M.E.; Levicán, G.; Ridley, C.; Díez, B. Distribution and Activity of Sulfur-Metabolizing Bacteria along the Temperature Gradient in Phototrophic Mats of the Chilean Hot Spring Porcelana. Microorganisms 2023, 11, 1803. https://doi.org/10.3390/microorganisms11071803
Konrad R, Vergara-Barros P, Alcorta J, Alcamán-Arias ME, Levicán G, Ridley C, Díez B. Distribution and Activity of Sulfur-Metabolizing Bacteria along the Temperature Gradient in Phototrophic Mats of the Chilean Hot Spring Porcelana. Microorganisms. 2023; 11(7):1803. https://doi.org/10.3390/microorganisms11071803
Chicago/Turabian StyleKonrad, Ricardo, Pablo Vergara-Barros, Jaime Alcorta, María E. Alcamán-Arias, Gloria Levicán, Christina Ridley, and Beatriz Díez. 2023. "Distribution and Activity of Sulfur-Metabolizing Bacteria along the Temperature Gradient in Phototrophic Mats of the Chilean Hot Spring Porcelana" Microorganisms 11, no. 7: 1803. https://doi.org/10.3390/microorganisms11071803